DOI:
10.1039/C6RA11110F
(Paper)
RSC Adv., 2016,
6, 62478-62484
Supramolecular prodrug micelles based on the complementary multiple hydrogen bonds as drug delivery platform for thrombosis therapy†
Received
29th April 2016
, Accepted 23rd June 2016
First published on 24th June 2016
Abstract
To improve the effect of thrombosis therapy, an amphiphilic supramolecular prodrug consisting of diosgenin derivative (theophylline–diosgenin) and uracil-terminated poly(ethylene glycol) (PEG-U) was designed and synthesized successfully. The prodrug could self-assemble into micelles which was not only enhanced the drug solubility but also prolonged systemic circulation. Bleeding time assay confirmed that the micelles have a better antithrombotic activity compared to diosgenin in vivo, and platelet aggregation assay in vitro got the same results. The low cytotoxicity of supramolecular copolymer micelles was confirmed by MTT assay against LO2/HK-2 cells and acute toxicity studies in mice. Based on these results, the supramolecular prodrug micelles are very promising candidates for thrombosis therapy.
Introduction
Thrombosis is one of the leading causes of morbidity and mortality throughout the world.1–6 To prevent and control the thrombogenic state, some anti-thrombotic drugs, such as anticoagulants, anti-platelet drugs and thrombolytic drugs, have been used in clinical application. However, most of the drugs exert many side effects, such as bleeding risk,7 narrow therapeutic window and incidence of resistance. Therefore, development of new drug candidates with minimal side effects and high potential for treatment of ischemic symptoms is urgently needed.8,9
In the process of searching for new antithrombotic drugs, traditional Chinese medicine have aroused people's attention.10,11 Dioscorea zingiberensis C. H. Wright (DZW), one of important Chinese medicinal materials, has been extensively used as a treatment of various diseases.12,13 More importantly, total steroidal saponins and diosgenin of Dioscorea zingiberensis C. H. Wright have been found to exert antithrombotic effect.14,15 Drugs with steroidal saponins being the main bioactive ingredients, such as Dioscorea Panthaica Prain et Burkill and Xue-sai-tong, have become a new commercial product for thrombosis therapy in China.16–18 Our group have been engaged in investigating the extraction and applications of steroidal saponins from D. zingiberensis for many years19,20 and have found that diosgenin showed a better antithrombotic activity through structural modification.21 Inspired by these researches, it can be inferred that conjugating diosgenin with other potential antithrombotic drugs to obtain diosgenin derivatives will be very promising for thrombosis therapy. However, due to the strong hydrophobicity of diosgenin, the diosgenin derivatives also suffer from limitations of poor physicochemical properties. Therefore, to fully exploit the potential of diosgenin derivatives, it is essential to address these issues.
To date, nanocarriers have attracted considerable attention for their application in drug delivery systems. Especially, polymeric micelles have emerged as one of the most promising drug delivery systems22–27 because of their advantages in increasing drug solubility, prolonging circulation time28,29 and improving pharmacokinetic properties.26–31 In the polymeric micelles systems, hydrophobic drug is incorporated in a polymeric matrix through chemical conjugation or physical entrapment, while the hydrophilic shell maintains a hydration barrier to provide a stable interface between the hydrophobic core and the external medium. Therefore, polymer micelles have great potential to serve as an efficient drug delivery system.
It is reported that xanthine drugs have potential antithrombotic effect.30,31 Therefore, in the present work, we designed and synthesized a novel antithrombotic prodrug of diosgenin and theophylline which is one kind of xanthine drugs (Scheme 1A). Furthermore, to enhance the solubility, bioavailability and pharmacokinetics of the drug, a hydrophilic polymer based on uracil and PEG was developed in this research. Theophylline and uracil can direct interaction by multiple hydrogen bonds and hydrophilic PEG-U was employed to construct supramolecular amphiphilic copolymers (D–T:U-PEG). Benefiting from their amphiphilicity and noncovalent connection, D–T:U-PEG could self-assemble into micelles with hydrophilic PEG shells and hydrophobic D–T cores in aqueous solution (Scheme 1B). The supramolecular prodrug micelles could increase the solubility of drug, prolong its systemic circulation in blood and improve pharmacokinetic properties, which is crucial for improving the drug efficacy in thrombosis therapy.
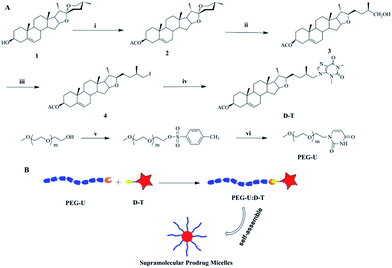 |
| | Scheme 1 (A) Schematic route of diosgenin derivative D–T and PEG-U. (i) Ac2O, pyridine, 60 °C, 2 h, 95%; (ii) AcOH, NaCNBH4, 35 °C, 2 h, 85%; (iii) NIS, Ph3P, CH2Cl2, 25 °C, 24 h, 95%; (iv) theophylline, K2CO3, DMF, 80 °C, 8 h, 75%. (v) NaOH, TSCl CH2Cl2, 0 °C, 48 h, 95%; (vi) K2CO3, uracil, DMF, 90 °C, 24 h, 65%. (B) Schematic illustration of the supramolecular prodrug self-assemble into micelles. | |
Materials and methods
Materials
N,N-Dimethylformamide (DMF), azabenzene (C5H5N), and methylene dichloride (CH2Cl2). Diosgenin (99%, Adams), acetic anhydride (99%, Adams), triethylamine (TEA, 99%, Adams), sodium cyanoborohydride (99%, Sigma), triphenylphosphine (99%, Sigma), n-iodosuccinimide (99%, Sigma), theophylline (99%, Sigma), uracil (99%, Sigma) and acetic acid (99%, J&K) were all used as received. Monomethoxy poly(ethylene glycol) (PEG) with Mn = 2000 g mol−1 was purchased from Fluka and dried by azeotropic distillation in the presence of dry toluene. Different polystyrene plates including 6- and 96-well ones were provided by Chinese Sangon Biotech. Male Balb/C mice (four weeks old, 18–22 g, Chengdu, China) and male Wistar rats (eight weeks old, 200–250 g, Chengdu, China) were used in this study.
Synthesis of 3β-hydroxy-(25R)-spirost-5-en-3β-acetate (2)
Diosgenin (8.3 g, 20 mmol) was taken in pyridine (100 mL) at room temperature and added acetic anhydride (20 mL, 213 mmol) to it at ice bath, then warmed to 60 °C. After the reaction, usual work-up was done as per reported method.32 Yield: 95%. 1H NMR (400 MHz, CDCl3) δ 5.37 (d, J = 4.5 Hz, 1H), 4.78–4.49 (m, 1H), 4.41 (dd, J = 14.9, 7.5 Hz, 1H), 3.47 (dd, J = 10.0, 3.2 Hz, 1H), 3.37 (t, J = 10.9 Hz, 1H), 2.48–2.23 (m, 2H), 2.03 (s, 3H), 1.03 (s, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.79 (d, J = 3.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 69.50, 138.67, 121.35, 108.25, 79.79, 72.87, 65.82, 61.07, 55.41, 48.92, 40.59, 39.24, 38.71, 37.07, 35.94, 35.71, 31.02, 30.82, 30.39, 29.28, 27.79, 26.72, 20.41, 19.79, 18.31, 16.12, 15.27, 13.51; ESI mass (MeOH): 457.3 [M + H]+, 479.3 [M + Na]+, 495.4 [M + K]+.
Synthesis of (25R) furost-5-en-,3β-acetoxy,26-ol (3)
Compound 2 (2.28 g, 5 mmol) was dissolved in a mixture solution of CH3COOH/CH2Cl2 (30 mL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 mL) at room temperature, and sodium cyanoborohydride (942 mg, 15 mmol) was added in portions over a period of 30 min. After 2 h, when the reaction was complete, 10 mL of ice-cool water poured the reaction mixture, extracted with ethyl acetate (3 × 40 mL), washed with 5% of sodium chloride solution and dried over anhydrous sodium sulphate. The organic layer was removed by vacuum distillation with a rotary evaporator to get a crude mass, which was purified through column chromatography over silica gel using petroleum ether–ethyl acetate as eluants. The desired alcohol 3 was obtained at 10–12% ethyl acetate–petroleum ether as white crystalline solid. Yield: 85%, 1H NMR (400 MHz, CDCl3) δ 5.37 (d, J = 4.9 Hz, 1H), 4.60 (tt, J = 10.5, 5.1 Hz, 1H), 4.31 (dd, J = 13.1, 7.7 Hz, 1H), 3.49 (dd, J = 10.6, 6.2 Hz, 1H), 3.43 (dd, J = 10.6, 6.2 Hz, 1H), 3.33 (td, J = 8.3, 3.8 Hz, 1H), 2.36–2.27 (m, 2H), 2.03 (s, 3H), 1.03 (s, 3H), 1.00 (d, J = 6.7 Hz, 3H), 0.91 (d, J = 6.7 Hz, 3H), 0.81 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 170.54, 139.67, 122.38, 90.35, 83.21, 73.89, 68.01, 65.09, 56.90, 50.00, 40.69, 39.40, 38.09, 37.92, 36.99, 36.70, 35.74, 32.22, 31.98, 31.56, 30.46, 30.13, 27.74, 21.42, 20.64, 19.32, 18.92, 16.63, 16.44; HR-MS (m/z) calcd for C29H46O4: 481.3294 [M + Na]+, found: 481.3283 [M + Na]+.
:15 mL) at room temperature, and sodium cyanoborohydride (942 mg, 15 mmol) was added in portions over a period of 30 min. After 2 h, when the reaction was complete, 10 mL of ice-cool water poured the reaction mixture, extracted with ethyl acetate (3 × 40 mL), washed with 5% of sodium chloride solution and dried over anhydrous sodium sulphate. The organic layer was removed by vacuum distillation with a rotary evaporator to get a crude mass, which was purified through column chromatography over silica gel using petroleum ether–ethyl acetate as eluants. The desired alcohol 3 was obtained at 10–12% ethyl acetate–petroleum ether as white crystalline solid. Yield: 85%, 1H NMR (400 MHz, CDCl3) δ 5.37 (d, J = 4.9 Hz, 1H), 4.60 (tt, J = 10.5, 5.1 Hz, 1H), 4.31 (dd, J = 13.1, 7.7 Hz, 1H), 3.49 (dd, J = 10.6, 6.2 Hz, 1H), 3.43 (dd, J = 10.6, 6.2 Hz, 1H), 3.33 (td, J = 8.3, 3.8 Hz, 1H), 2.36–2.27 (m, 2H), 2.03 (s, 3H), 1.03 (s, 3H), 1.00 (d, J = 6.7 Hz, 3H), 0.91 (d, J = 6.7 Hz, 3H), 0.81 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 170.54, 139.67, 122.38, 90.35, 83.21, 73.89, 68.01, 65.09, 56.90, 50.00, 40.69, 39.40, 38.09, 37.92, 36.99, 36.70, 35.74, 32.22, 31.98, 31.56, 30.46, 30.13, 27.74, 21.42, 20.64, 19.32, 18.92, 16.63, 16.44; HR-MS (m/z) calcd for C29H46O4: 481.3294 [M + Na]+, found: 481.3283 [M + Na]+.
Synthesis of (25R) furost-5-en-,3β-acetoxy,26-theophylline (D–T)
N-Iodosuccinimide (NIS, 490 mg, 2.2 mmol) was dissolved in a 10 mL methylene dichloride at room temperature. Then, the methylene dichloride solution of triphenylphosphine (500 mg, 2.2 mmol) was added dropwise into this solution. After 10 min, compound 3 (500 mg, 1.1 mmol) was added into above mixed solution and stirred for an additional 24 h at room temperature. The resulting mixture washed with water and dried over anhydrous sodium sulphate. The organic layer was removed by vacuum distillation with a rotary evaporator to get a yellow oily substance, which was purified through column chromatography over silica gel using petroleum ether–ethyl acetate as eluants. Compound 4 was obtained with a yield of 95%. After that, compound 4 (1 g, 1.76 mmol), theophylline (634 mg, 3.52 mmol) and potassium carbonate (486 mg, 3.52 mmol) were dissolved in a 20 mL N,N-dimethylformamide and stirred for an additional 8 h at 80 °C. The resulting mixture was filtered and filtrate was removed by evaporation to get a yellow solid substance, which was purified through column chromatography over silica gel using petroleum ether–ethyl acetate as eluants. The desired D–T was obtained at 50% ethyl acetate–petroleum ether as white solid. Yield: 75%, 1H NMR (400 MHz, CDCl3) δ 7.52 (1H, –NCHN–), 5.37 (1H, –CCHCH2–), 4.60 (1H, –CH2CH(O)CH2–), 4.29 (1H, –CHCH(CH2)O–), 4.19 (1H, –CHCH(CH2)O–), 4.12–4.01 (2H, –CHCH2N–), 3.60 (3H, –CH3), 3.41 (3H, –CH3), 3.27 (2H, –CHCH2C–), 2.03 (3H, CH3–), 1.04 (3H, CH3–), 0.97 (3H, CH3–), 0.91 (3H, CH3–), 0.79 (3H, CH3–); 13C NMR (101 MHz, CDCl3) δ 170.54, 155.17, 151.68, 149.12, 148.87, 141.43, 139.69, 122.35, 107.10, 90.06, 83.24, 77.37, 77.26, 77.06, 76.74, 73.87, 64.95, 56.86, 53.16, 49.96, 40.68, 39.37, 38.08, 37.88, 36.98, 36.70, 34.51, 32.19, 31.97, 31.55, 31.01, 30.35, 29.78, 28.00, 27.74, 21.44, 20.63, 19.34, 18.79, 16.88, 16.45. HR-MS (m/z) calcd for C36H52N4O5: 621.4018 [M + H]+, 643.3836 [M + Na]+, found: 621.4017 [M + H]+, 481.3842 [M + Na]+.
Preparation of PEG-U
PEG (6.0 g, 1.2 mmol) and NaOH (0.14 g, 3.36 mmol) in anhydrous CH2Cl2 (50 mL) were stirred in an ice water bath. After stirring for 40 min, a CH2Cl2 solution (20 mL) of p-toluenesulfonyl chloride (TsCl; 0.41 g, 2.2 mmol) was added dropwise into the reaction mixture and reacted at 0 °C for 48 h. After reaction, 10 mL of water was poured into the resulting mixture and extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried with anhydrous sodium sulfate (Na2SO4), and filtered, then, the filtrate was evaporated. PEG–sulfanilic acid ester was obtained with a yield of 95%. After that, uracil (0.36 g, 3.2 mmol), PEG–sulfanilic acid ester (8.0 g, 1.6 mmol), potassium carbonate (K2CO3, 0.44 g, 3.2 mmol) were dissolved in 100 mL of DMF and stirred at 90 °C for 24 h. After reaction, 100 mL of water was poured the reaction mixture and extracted with CH2Cl2 (3 × 50 mL). After drying with anhydrous sodium sulfate (Na2SO4), most of the solvent was removed by evaporation. The residue was precipitated into cold ethanol and dried under vacuum to a constant weight. Yield: 65%. 1H NMR (400 MHz, CDCl3, 298 K) δ ppm: 7.7 (1H, –CONHCO–), 7.3 (1H, –NCHCH–), 5.6 (1H, –CHCHCO–), 3.9 (2H, –CH2CH2N–), 3.5–3.7 (PEG, 2H per repeating unit, –OCH2CH2–, 2H per repeating unit, –O–CH2CH2–), 3.3 (3H, CH3– OCH2–). 13C NMR (100 MHz, CDCl3, 298 K) δ: 47.04 (–CH2CH2N–), 58.0 (CH3O–), 67.8 (–CH2CH2N–), 69.7 (–CH2CH2O–), 71.2 (CH3OCH2–), 100.2 (–CHCHCO–), 146.3 (–CHCHCO–), 150.9 (–NCONH–), 162.4 (–CHCONH–). HR-MS (m/z, Fig. S7†): 2131.2563 [M + Na]+.
Formation of the supramolecular prodrug micelles
A typical procedure to obtain the supramolecular prodrug micelles is as follows: at room temperature, both D–T (3.1 mg, 0.005 mol) and PEG-U (10 mg, 0.005 mol) were dissolved in THF (1 mL). After that, the polymer solution was put dropwise into 10 mL of deionized water and stirred slightly for 40 min. Then the THF was removed by vacuum distillation. The micelles solution was dialyzed against deionized water for 48 h (MWCO = 2000), during which the water was renewed every 4 h.
Dynamic light scattering (DLS) and transmission electron microscopy (TEM)
Dynamic light scattering (DLS) measurements were performed to determine the micellar size and distribution using a Malvern Zetasizer Nano Series equipped with DTS software and operating a 4 mW He–Ne laser at 633 nm. All samples were tested at different temperature (from 25 °C to 90 °C) and a scattering angle of 90°. TEM tests were obtained using a JEOL JEM-100CX-II instrument at a voltage of 200 kV. Samples were prepared by drop-casting nano-aggregate solutions onto carbon-coated copper grids and then freeze drying under vacuum before the measurements.
Bleeding time assay
Male Balb/C mice were randomly divided into three groups of ten each. Before the bleeding time was assessed, the mice were given diosgenin and control buffer orally in a volume of 200 μL, or other mice were given the supramolecular prodrug micelles with intraperitoneal in a volume of 200 μL twice a day for five days. The bleeding time was measured by the method of Nobukata33 with slight modification. In brief, an incision was made at the tip of the tail of anesthetized mouse with a scalpel, and the tail was immersed in normal saline (37 °C) immediately. The time from the incision to the cessation of bleeding was recorded. A bleeding time of 900 s was used as the cut-off time for the purpose of statistical analysis. The measurement of bleeding time was performed in a blinded fashion.
Blood preparation
For in vitro platelet aggregation, Wistar rats (200–250 g) were divided randomly into three groups of ten animals each. After five days, rats were anesthetized with i.p. pentobarbital sodium (40 mg kg−1), and whole blood was collected by cardiac puncture and anti-coagulated with 3.8% trisodium citrate (9:1, v/v). Platelet-rich plasma (PRP) was obtained by centrifugation at 100g for 10 min. Platelet-poor plasma (PPP) was prepared by centrifugation at 1000g for 10 min.
Platelet aggregation assay in vitro
The platelet aggregation assay was performed at 37 °C using a LBY-NJ4 aggregometer (Pulisheng, Beijing, China). PRP were preincubated with the indicated doses of diosgenin and the supramolecular prodrug micelles, or control buffer for 40 min at 37 °C under static conditions. Aggregation was induced by 20 μM ADP, and platelet aggregation in vitro was measured.
Cell culture
The cultured human hepatocyte LO2 cell line and human kidney HK-2 cell line were provide by Laboratory of Transplantation Immunity, SiChuan University and grown in DMEM (HyClone, USA) supplemented with 10% FBS (HyClone, USA), 100 units per mL penicillins and 100 μg mL−1 streptomycin as previously described. Cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C.
LO2 cells and HK-2 cells were cultured in complete medium with 10% FBS to 80% cell confluence and subjected to assays after overnight serum depletion. The supramolecular prodrug micelles and diosgenin dissolved in DMSO were added to the medium. The final concentration of DMSO did not exceed 0.1%, which did not affect cell viability.
Cytotoxicity measurements of the supramolecular prodrug micelles and diosgenin in vitro
LO2 cells and HK-2 cells were seeded into 96-well plates (Corning, USA) at a density of 3 to 5 × 103 cells per well and incubated overnight, then exposed to various concentrations of micelles and diosgenin dissolved in DMSO and diluted with DMEM medium for 24 h. The cytotoxicity of samples in 0.1% DMSO (final DMSO concentration in medium) was tested using MTT assay and images were taken by an optical microscope (Olympus BX51, Japan) after 24 h of incubation. In brief, cells were washed once with PBS carefully, incubated with 0.2 mL of serum-free DMEM medium containing 0.05% MTT for 4 h. After that, the culture medium was removed and 0.15 mL of DMSO was added to solubilise the formed formazan. The absorbance of each well was measured at 490 nm with a microplate reader (Molecular Devices, USA). We compared the absorbance of the treated cells with the control cells, which were considered as the 100% viability value.
Acute toxicity studies in mice
The toxicity of diosgenin and the supramolecular prodrug micelles to healthy male Kunming mice was conducted by oral administration of diosgenin (200 and 400 mg kg−1) and the supramolecular prodrug micelles with intraperitoneal injection (50 and 100 mg kg−1). Saline was injected as control. After one week, major organs (heart, liver, spleen, lung, kidney and brain) and blood samples from each mouse were harvested. Viscera samples were fixed in formalin, processed routinely into paraffin, sectioned, stained with hematoxylin and eosin (H&E) (Aladdin Chemical Co., Ltd., Shanghai, China) and examined by an optical microscope (Olympus Corporation, Tokyo, Japan). Serum was collected to measure alanine transaminase (ALT), aspartate transaminase (AST).
Result and discussion
Synthesis and characterization of D–T and PEG-U
The synthetic route of D–T and PEG-U is outlined in Scheme 1A. D–T was synthesized by substitution reaction with a molar feed ratio of compound 4/theophylline at 1:2. The potassium carbonate catalytic method was used to make compound 4 react with theophylline to produce the D–T. The chemical structure of D–T was confirmed by 1H and 13C nuclear magnetic resonance spectroscopy (1H NMR, 13C NMR). Typical 1H NMR and 13C NMR spectra of D–T is shown in Fig. 1A and S4.† For the 1H NMR, the peak at 7.52 (1H, –NCHN–), 3.60 (3H, –CH3) and 3.41 (3H, –CH3) belong to theophylline, and 5.37 (1H, –CCHCH2–), 4.60 (1H, –CH2CH(O)CH2–), 4.29 (1H, –CHCH(CH2)O–), 4.19 (1H, –CHCH(CH2)O–) belong to diosgenin. The structure of the product was confirmed by these detailed data of the chemical shifts. The 13C NMR also further demonstrate the successful conjugating diosgenin with theophylline. The LC and HRMS techniques were used to characterize the purity and molecular weight of D–T and the results are shown in S5 and S6.† The LC profile gives the retention time of D–T as 2.42 min, indicating the high purity of D–T. The HRMS data show that the molecular weight of D–T is (m/z, M + H+) 621.4017, which is consistent with the calculated value (m/z, M + H+, 621.4018).
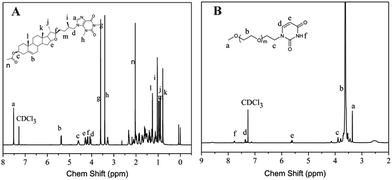 |
| | Fig. 1 1H NMR spectra of diosgenin derivative (D–T) and uracil-terminated poly(ethylene glycol) (PEG-U): (A) D–T, (B) PEG-U (400 MHz, CDCl3). | |
PEG-U was synthesized by the reaction of PEG–sulfanilic acid ester with excess uracil. The synthetic route is shown in Scheme 1B. The chemical structure identification data of PEG-U was provided by 1H NMR and 13C NMR spectroscopy. As shown in Fig. 1B, for the 1H NMR, the peaks at 3.9 and 7.7 ppm attribute to the protons of CH2 (–CH2CH2N–) connected with uracil and NH (–CONHCO–) on the uracil, which demonstrates the generation of PEG-U. The 13C NMR (Fig. S8†) also further demonstrate the successful grafting of uracil on PEG.
Formation of micelles
The theophylline in D–T and uracil in PEG-U can direct interaction by multiple hydrogen bonds to form supramolecular copolymer. Since the PEG-U is a water-soluble compound, D–T is a water-insoluble drug. Therefore, the supramolecular copolymer (D–T:U-PEG) is amphiphilic. Benefiting from this amphiphilic nature, the supramolecular copolymer could self-assemble to form micelles in aqueous solution. The supramolecular micelles prodrug were prepared by dialysis method. After adding the THF solution of D–T and PEG-U into deionized water, the THF was removed by dialysis and a stable nanoparticle solution with a concentration of 1 mg mL−1 was obtained. Further, to confirm that the supramolecular prodrug can form nanoparticles, both DLS and TEM measurements were used to study the size and morphology of the supramolecular prodrug micelles. As shown in Fig. 2A, the supramolecular prodrug micelles aqueous solution forms aggregates and the mean hydrodynamic diameter of the supramolecular prodrug micelles aggregates is about 187.2 nm with a unimodal size distribution. TEM technique was used to further measure and visualize the size and morphology of the aggregates. The TEM image in Fig. 2B showed that the supramolecular prodrug micelles aggregated into approximate spherical micelles in water and the micelles sizes determined by TEM were in accordance with the data from DLS in Fig. 2A. These results demonstrated that the supramolecular prodrug can self-assemble into stable and well-defined nanoparticles.
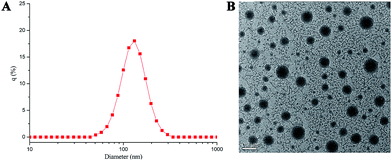 |
| | Fig. 2 (A) DLS results for prodrug micelles; (B) TEM photograph of prodrug micelles. | |
Acidic and thermal stability of the supramolecular prodrug micelles
The hydrogen bonding interactions between hydrophobic D–T and hydrophilic PEG-U made copolymer micelles unstable at acidic pH. To study the ability of the micelles in physiological condition, the micelles were treated with pH 7.4 PBS buffer and pH 5.0 acetate buffer (50 mM). The particle sizes were measured by DLS at different time intervals. The result in Fig. 3A showed that the supramolecular prodrug micelles aggregate rapidly at pH 5.0. The mean diameter of the copolymer micelles increases from 187.2 nm to about 520.9 nm in 4 h, while it doesn't change in PBS buffer (pH = 7.4). The stability of prepared micelles under physiological pH condition (pH 7.4) provides the possibility for the introduction to the body with the potential of therapeutics delivery. The driving force for the change of micelles size at low pH value is attributed to the protonation of the nucleobase nitrogen atoms, which leads to the shedding of hydrophilic PEG shell from the micelles and the aggregation of hydrophobic D–T core.34
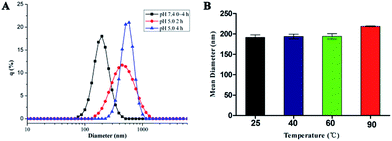 |
| | Fig. 3 (A) Size change of prodrug micelles at different pH over time at 25 °C monitored by DLS. (B) The size of prodrug micelles at different temperatures determined by DLS. Error bars represent the standard deviation (n = 3). | |
The thermal stability of supramolecular copolymers was investigated at different temperatures. Fig. 3B showed that the effect of temperature on the supramolecular prodrug micelles was very slight at the temperature below 60 °C and the mean diameter is around 187.2 nm. When the temperature was up to 90 °C, the size of micelles was increased to 217.9 nm because of the dissociation of complementary hydrogen bonds at high temperature. The stability of prepared micelles at physiological temperature provided the possibility for the introduction to the body with the potential of therapeutics delivery.
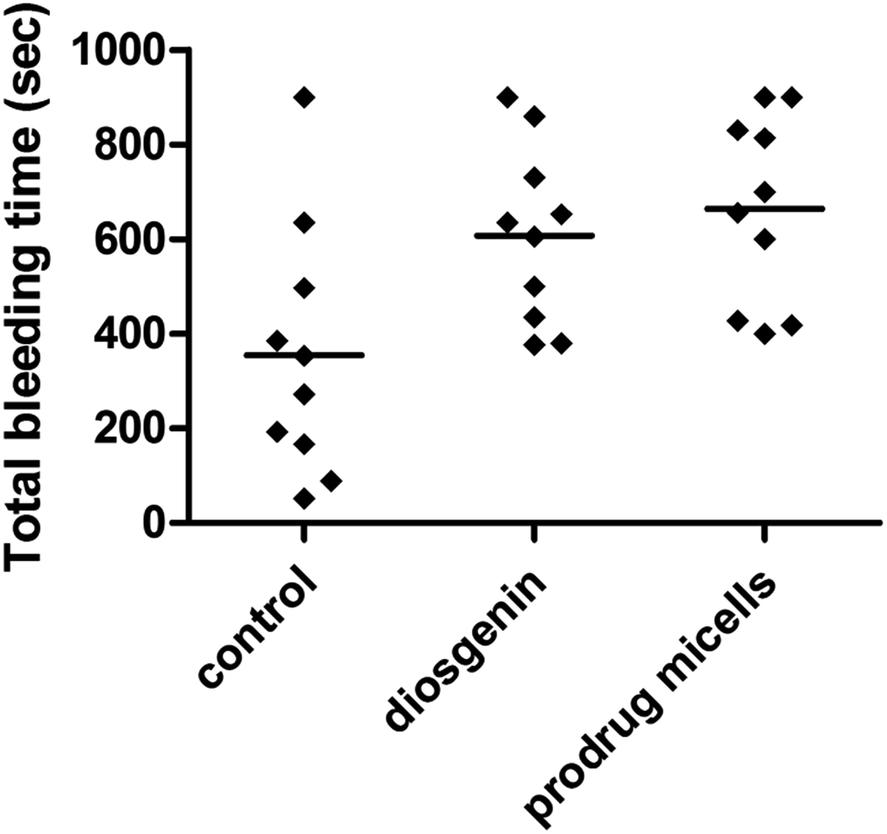
Bleeding time in mice
To investigate the effect of diosgenin and the supramolecular prodrug micelles on normal thrombosis, we measured bleeding time one and a half-hour after the final oral administration of diosgenin (100 mg kg−1) and the prodrug micelles with intraperitoneal injection (50 mg kg−1). As shown in Fig. 4, both diosgenin and prodrug micelles showed antithrombotic effects compared to controls, and prodrug micelles exhibited more outstanding efficiency in prolonging bleeding time than controls and diosgenin. The mean bleeding time for the prodrug micelles and diosgenin treatment was 629.4 s and 605.0 s. It can be drawn from the above data that the prodrug micelles have much better physicochemical properties compared to diosgenin, and can improve the diosgenin's drug efficacy in thrombosis therapy.
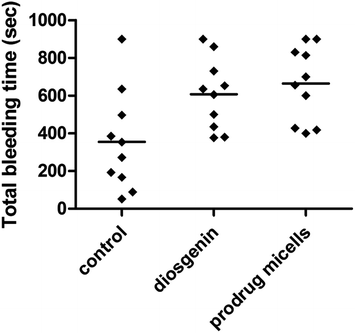 |
| | Fig. 4 Effect of diosgenin and prodrug micelles on bleeding time. The tail bleeding time in mice given diosgenin and prodrug micelles, or saline was measured as described in experiment section. | |
Platelet aggregation assay in vitro
To investigate whether the supramolecular prodrug micelles have better antithrombotic effect than diosgenin in vitro. We examined the prodrug micelles and diosgenin directly to inhibit platelet aggregation in vitro. As shown in Fig. 5, the results indicated that the prodrug micelles exhibited more outstanding efficiency in inhibiting rat platelet aggregation than diosgenin in vitro. At same time, the results also indicated that the micelles inhibited rat platelet aggregation in a dose-dependent manner. As known to all, hemostasis has three major steps: (1) vasoconstriction, (2) formation of a platelet plug by platelet aggregation and (3) blood coagulation. Therefore, the measurement of bleeding time and platelet aggregation can comprehensively reflect the anti-thrombotic activities of drugs. In other words, bleeding time and platelet aggregation will change if anti-platelet aggregation drugs are taken up by platelet. As shown in Fig. 4 and in 5, the results indicated that the prodrug micelles have better antithrombotic effect than diosgenin in vivo and in vitro. Videlicet, the results indicated that the prodrug micelles can be better taken up than diosgenin by platelet. At the same time, because the prodrug micelles can release the prodrug in an acidic environment, the prodrug micelles can release the prodrug in the platelet and exert antithrombotic effect in vivo and in vitro.
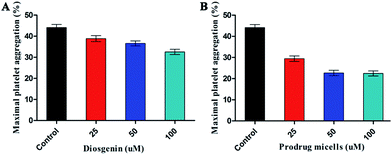 |
| | Fig. 5 (A) Effect of diosgenin and (B) effect of prodrug micelles on platelet aggregation in rats. | |
Cytotoxicity of the supramolecular prodrug micelles in vitro
In vitro cell culture systems are a useful tool to rapidly assess the potential safety or toxicity of chemical constituents. Liver and kidney, representing the major metabolism, bioactivation and elimination organ respectively, are the first choice in assessment of the safety/toxicity of chemical constituents. Here, we investigated the effect of the prodrug micelles and diosgenin on LO2 cells and HK-2 cells viability by using MTT assay. As shown in Fig. 6, the varying concentrations from 2.5 to 50 μM L−1 of the prodrug micelles did not influence LO2 and HK-2 cell survival after 24 h exposure. The 2.5 to 50 μM L−1 of the prodrug micelles stimulated the cell proliferation without a significant morphological change (Fig. S9†). However, the cytotoxicity of diosgenin to LO2 and HK-2 cell is higher than the prodrug micelles at varying concentrations from 2.5 to 50 μM L−1. These results suggest that the prodrug micelles have low cytotoxicity and are safe for LO2 and HK-2 cell using concentrations in the experiment.
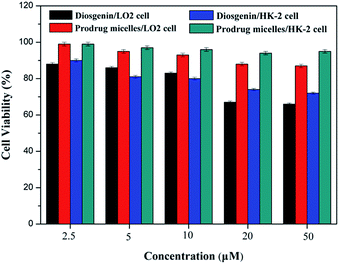 |
| | Fig. 6 In vitro toxicity of diosgenin and prodrug micelles to LO2 and HK-2 cells after 24 h incubation at different concentrations. | |
Acute toxicity of the supramolecular prodrug micelles studies in mice
In the acute toxicity study, no cell death was found in any of the animals injected through intraperitoneal with the prodrug micelles (50 and 100 mg kg−1 body weight) and conducted by oral administration of diosgenin (200 and 400 mg kg−1). As shown in Fig. 7, the histological studies revealed that there was no significant histological alterations in all major organs (heart, liver, spleen, lung, kidney and brain) of mice treated with the prodrug micelles and diosgenin in comparison with control group. These results verified that no observable toxicity in tissue level of mice was induced by the prodrug micelles, suggesting good biocompatibility of the prodrug micelles. In addition, Table 1 shows that no significant difference was observed in the levels of ALT and AST in the serum of mice treated with various doses of the prodrug micelles and diosgenin, further confirming no hepatotoxicity of the prodrug micelles.
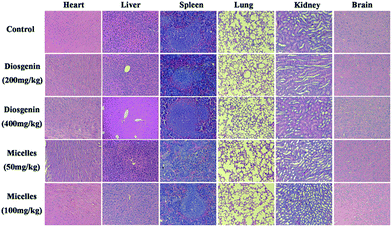 |
| | Fig. 7 Hematoxylin-eosin staining after one week of prodrug micelles given intraperitoneal injecting and diosgenin given oral administration at different dose in mice. | |
Table 1 Effects of the diosgenin and prodrug micelles on AST and ALT levels in the serum of mice
| Group |
AST (U L−1) |
ALT (U L−1) |
| Control |
104.56 ± 0.73 |
28.37 ± 0.87 |
| 200 mg kg−1 of diosgenin |
117.23 ± 6.17 |
38.53 ± 2.38 |
| 400 mg kg−1 of diosgenin |
115.60 ± 3.72 |
30.13 ± 0.61 |
| 50 mg kg−1 of prodrug micelles |
109.80 ± 2.34 |
31.27 ± 0.82 |
| 100 mg kg−1 of prodrug micelles |
108.30 ± 1.65 |
29.63 ± 0.87 |
Conclusions
In this study, a novel supramolecular prodrug micelles based on the complementary multiple hydrogen bonds of nucleobases was prepared and used for thrombosis therapy. Moreover, the supramolecular prodrug micelles could self-assemble into micelles, in which diosgenin derivative was located in the hydrophobic part and MPEG was the hydrophilic corona. The drug delivery system increased the solubility and reduced side effects, leading to a better efficacy of thrombus than the diosgenin. Overall, we believe that the amphiphilic supramolecular drug micelles may open a new strategy to improve solubility and bioavailability of drug and are very promising candidates for improvements in drug efficacy.
Acknowledgements
This study was partly supported by the China National ‘12.5’ Foundation (Grant No. 2011BAJ07B04) and National Natural Science Foundation of China (Grant No. 20972105, 51503130).
Notes and references
- J. B. Yang, G. Q. Su, Y. Ren and Y. Chen, Bioorg. Med. Chem. Lett., 2015, 25, 492–495 CrossRef CAS PubMed.
- M. Y. Wu, D. D. Wen, N. Gao, C. Xiao, L. Yang, L. Xu, W. Lian, W. L. Peng, J. M. Jiang and J. H. Zhao, Eur. J. Med. Chem., 2015, 92, 257–269 CrossRef CAS PubMed.
- X. Y. Zhang, W. Wang, S. L. Cheng, M. Zhao, M. Q. Zheng, H. W. Chang, J. H. Wua and S. Q. Peng, Eur. J. Med. Chem., 2010, 18, 1536–1554 CAS.
- M. Q. Zheng, X. Y. Zhang, M. Zhao, H. W. Chang, W. Wang, Y. J. Wang and S. Q. Peng, Eur. J. Med. Chem., 2008, 16, 9574–9587 CAS.
- N. Mackman, Nature, 2008, 451, 914–918 CrossRef CAS PubMed.
- M. G. Beckman, W. C. Hooper, S. E. Critchley and T. L. Ortel, Am. J. Prev. Med., 2010, 38, S495–S501 CrossRef PubMed.
- B. M. Massie, J. F. Collins, S. E. Ammon, P. W. Armstrong, J. G. F. Cleland, M. Ezekowitz, S. M. Jafri, W. F. Krol, C. M. O'Connor, K. A. Schulman, K. Teo and S. R. Warren, Circulation, 2009, 119, 1616–1624 CrossRef CAS PubMed.
- I. Melnikova, Nat. Rev. Drug Discovery, 2009, 8, 353–354 CrossRef CAS PubMed.
- U. Trstenjak, J. Ilaš and D. Kikelj, Eur. J. Med. Chem., 2013, 64, 302–313 CrossRef CAS PubMed.
- Y. Athukorala, K. W. Lee, S. K. Kim and Y. J. Jeon, Bioresour. Technol., 2007, 98, 1711–1716 CrossRef CAS PubMed.
- H. A. O. Rocha, F. A. Moraes, E. S. Trindade, C. R. C. Franco, R. J. S. Torquato, S. S. Veiga, A. P. Valente, P. A. S. Mourão, E. L. Leite, H. B. Nader and C. P. Dietrich, J. Biol. Chem., 2005, 280, 41278–41288 CrossRef CAS PubMed.
- S. G. Sparg, M. E. Light and J. V. Staden, J. Ethnopharmacol., 2004, 94, 219–243 CrossRef CAS PubMed.
- D. Z. Chen, Chinese virose plant, Science Press, Beijing, 1st edn, 1985 Search PubMed.
- G. H. Gong, Y. Qin and W. Huang, Phytomedicine, 2011, 18, 458–463 CrossRef CAS PubMed.
- H. Li, W. Huang, Y. Q. Wen, G. H. Gong, Q. B. Zhao and G. Yu, Fitoterapia, 2010, 81, 1147–1156 CrossRef CAS PubMed.
- X. M. Sun, Y. Yao, S. J. Ji and J. B. Chen, J. Math. Med., 2001, 14, 26–27 Search PubMed.
- The State Pharmacopoeia Commission of PR China, Pharmacopoeia of the People's Republic of China, Chemical Industry Press, Beijing, 2000, vol. 1 Search PubMed.
- Y. Qin, X. H. Wu, W. Huang, G. H. Gong, D. Li, Y. He and Y. L. Zhao, J. Ethnopharmacol., 2009, 126, 543–550 CrossRef CAS PubMed.
- Q. Y. Tong, Y. He, Q. B. Zhao, Y. Qing, W. Huang and X. H. Wu, Steroids, 2012, 77, 1219–1227 CrossRef CAS PubMed.
- G. H. Gong, Y. Qin, W. Huang, S. Zhou, X. H. Wu, X. H. Yang, Y. L. Zhao and D. Li, Chem.-Biol. Interact., 2010, 184, 366–375 CrossRef CAS PubMed.
- R. Zhang, B. Z. Huang, D. Dua, X. R. Guo, G. Xin, Z. H. Xing, Y. Liang, Y. N. Chen, Q. M. Chen, Y. He and W. Huang, Steroids, 2013, 78, 1064–1070 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- H. Arimura, Y. Ohya and T. Ouchi, Biomacromolecules, 2005, 6, 720–725 CrossRef CAS PubMed.
- Y. C. Wang, L. Y. Tang, T. M. Sun, C. H. Li, M. H. Xiong and J. Wang, Biomacromolecules, 2007, 9, 388–395 CrossRef PubMed.
- J. Y. Liu, Y. Pang, W. Huang, X. Y. Zhu, Y. F. Zhou and D. Y. Yan, Biomaterials, 2010, 31, 1334–1341 CrossRef CAS PubMed.
- J. Y. Liu, W. Huang, Y. Pang, X. Y. Zhu, Y. F. Zhou and D. Y. Yan, Langmuir, 2010, 26, 10585–10592 CrossRef CAS PubMed.
- A. Rosler, G. W. M. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95–108 CrossRef CAS PubMed.
- Y. N. Xue, Z. Z. Huang, J. T. Zhang, M. Liu, M. Zhang, S. W. Huang and R. X. Zhuo, Polymer, 2009, 50, 3706–3713 CrossRef CAS.
- Y. Q. Yang, L. S. Zheng, X. D. Guo, Y. Qian and L. J. Zhang, Biomacromolecules, 2010, 12, 116–122 CrossRef PubMed.
- J. S. Franzone, R. Cirillo, M. C. Reboani, M. V. Torrielli and L. M. Pernigotti, Drugs Exp. Clin. Res., 1988, 14(5), 347–354 CAS.
- CA: Vol 92, 174544v 1980.
- A. A. Hamid, M. Hasanain, A. Singh, B. Bhukya, Omprakash, P. G. Vasudev, J. Sarkar, D. Chanda, F. Khan, O. O. Aiyelaagbe and A. S. Negi, Steroids, 2014, 87, 108–118 CrossRef CAS PubMed.
- H. Nobukata, Y. Katsuki, T. Ishikawa, M. Inokuma and Y. Shibutani, Toxicol. Lett., 1999, 104, 93–101 CrossRef CAS PubMed.
- K. C. F. Leung, C. P. Chak, C. M. Lo, W. Y. Wong, S. Xuan and C. H. K. Cheng, Chem.–Asian J., 2009, 4, 364–381 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11110f |
| ‡ Wen'en Li and Haibo Wang contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.