
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree cyclic pentapeptides and a cyclic lipopeptide produced by endophytic Fusarium decemcellulare LG53†
Gang
Li
a,
Souvik
Kusari
 *a,
Christopher
Golz
b,
Carsten
Strohmann
b and
Michael
Spiteller
*a
*a,
Christopher
Golz
b,
Carsten
Strohmann
b and
Michael
Spiteller
*a
aInstitute of Environmental Research (INFU), Department of Chemistry and Chemical Biology, Chair of Environmental Chemistry and Analytical Chemistry, TU Dortmund, Otto-Hahn-Straße 6, 44221 Dortmund, Germany. E-mail: souvik.kusari@tu-dortmund.de; michael.spiteller@tu-dortmund.de; Fax: +49-231-755-4084; Fax: +49-231-755-4085; Tel: +49-231-755-4086 Tel: +49-231-755-4080
bInorganic Chemistry, Department of Chemistry and Chemical Biology, TU Dortmund, Otto-Hahn-Straße 6, 44221 Dortmund, Germany
First published on 3rd June 2016
Abstract
Three new cyclic pentapeptides (1–3) and a known cyclic lipopeptide, fusaristatin A (4), were isolated from an endophytic fungus, Fusarium decemcellulare LG53, harbored in a Chinese medicinal plant Mahonia fortunei. The planar structures of the new compounds were elucidated by extensive spectroscopic analyses using HRMSn and NMR. Their absolute configurations were unambiguously determined by single crystal X-ray diffraction and Marfey's method. Interestingly, compounds 1–3 exhibited no antibacterial activity against a panel of Gram-positive and Gram-negative bacteria. In order to explore the plausible ecological relevance of the compounds within the context of microbial crosstalk and communication strategies, we designed antagonistic and dual-culture assays combined with matrix-assisted laser desorption ionization imaging high-resolution mass spectrometry (MALDI-imaging-HRMS) to investigate the interaction of F. decemcellulare LG53 with another fungal endophyte Glomerella acutata LG52 isolated from the same tissue of the plant. Compound 4 was found to display an allelopathic effect on endophytic G. acutata LG52. Our results provide a proof-of-concept of balanced antagonism that might be occurring between endophytes inhabiting the same plant tissues in distinct ecological niches.
Introduction
Endophytic microorganisms, commonly known as ‘endophytes’, represent a colossally diverse group of organisms that reside within plant tissues in a broadly mutualistic fashion.1 The enormous biological diversity of endophytes coupled to their multifaceted interactions with associated organisms (e.g. other endophytes, invading pathogens, insects, pests and feeders) often leads towards the coevolution of specific functional traits. These traits range from synergism, physical and chemical neighbor communication, to allelopathy, which are a part of the overall mutualistic network of endophytes with associated macro- and microorganisms.1In our continuing efforts to isolate and characterize endophytes with desirable functional traits, we recently embarked on investigating the Chinese medicinal plant belonging to the Berberidaceae family, Mahonia fortunei2 prospected from different populations and locations. We isolated a plethora of culturable endophytic fungi from plants prospected from Shanghai and Guangdong, People's Republic of China. On employing the OSMAC (One Strain Many Compounds) approach3 on the endophytes isolated from the Shanghai plants, some interesting bioactive compounds were obtained.2
Herein we report the isolation and characterization of an endophytic fungus, Fusarium decemcellulare LG53, harboring the stem of a M. fortunei plant collected from Guangdong, People's Republic of China. Chemical investigation on this fungus after employing the OSMAC approach led to the discovery of three new cyclic pentapeptides (1–3) along with a known cyclic lipopeptide, fusaristatin A (4).4 The structures of isolated compounds 1–3 were elucidated by extensive spectroscopic analyses using HRMS, HRMSn, and NMR, and their absolute configurations were unambiguously determined by single crystal X-ray diffraction and Marfey's method. Following-up on one of our research strategies of traditional knowledge-based de-replication of the functional roles of endophyte isolates,2 the new compounds (1–3) were evaluated for in vitro antibacterial activities. Interestingly, none of them exhibited in vitro antibacterial activity against a panel of Gram-positive and Gram-negative bacteria. Furthermore, fusaristatin A (4), known not to possess antimicrobial efficacy,4–6 was also produced by the endophyte, which led us to the question the plausible ecological relevance of these compounds. Taking cues from our previous work on cyclopeptides functioning as microbial neighbor “crosstalk” molecules between an endophytic fungus and an associated bacterium,7 we embarked on co-culture investigation of F. decemcellulare LG53 with another fungal endophyte Glomerella acutata LG52 isolated from the same tissue of the plant using antagonistic and dual-culture assays coupled to matrix-assisted laser desorption ionization imaging high-resolution mass spectrometry (MALDI-imaging-HRMS). We discuss our results within the context of plausible chemical interactions occurring between coexisting endophytes in situ.1
Results and discussion
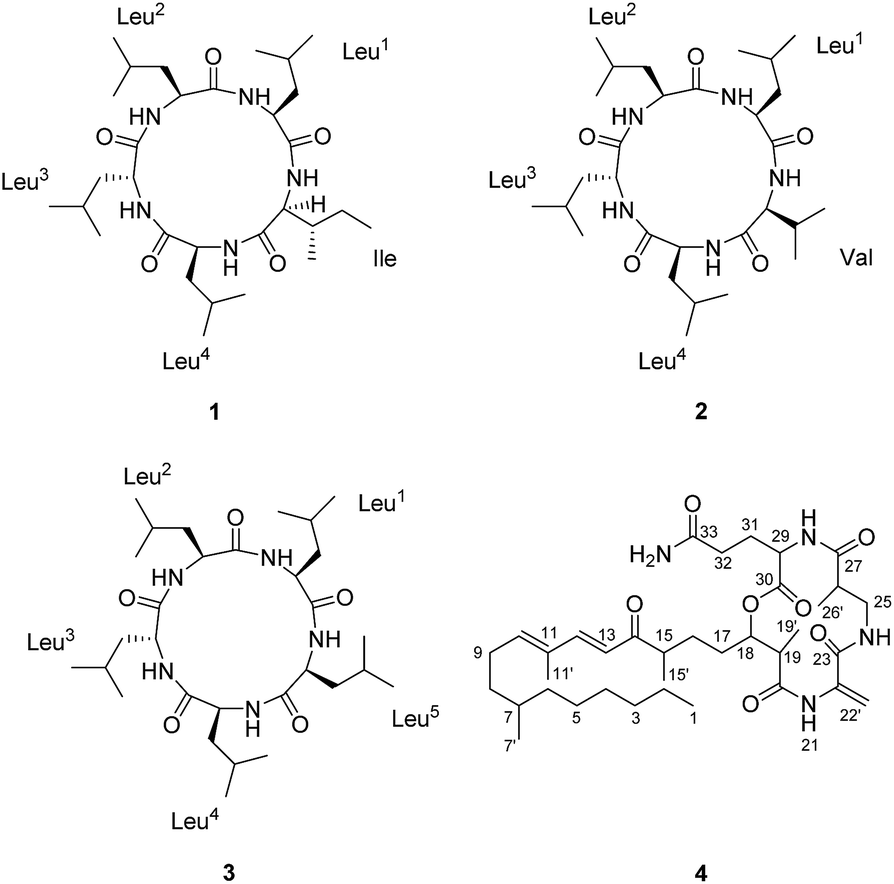
Compound 1 (Fig. 1) was isolated as a white amorphous powder with a molecular formula C30H55O5N5 (six double bond equivalents) as determined by ESI-HRMS (m/z 566.4282 [M + H]+, calcd 566.4276, Δ 1.1058 ppm; Fig. S1, ESI†). HR-CID (collision induced dissociation)-MS2 experiment (Fig. 2) showed three leucine/isoleucine (Leu/Ile) residues and an important fragment ion at m/z 227.1752 [M + H-3 × C6H11ON]+ (2 × Leu/Ile) from the pseudomolecular ion at m/z 566.43 [M + H]+, which was in accordance with the previously reported cyclo-(L-Leu-L-Leu-L-Leu-L-Leu-L-Ile).8 Firstly, we used CDCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD3OD (1:3) as the solvent for NMR measurements. The 1H and 13C NMR data (Fig. S3 and S4, ESI†) displayed the similar structural features as that for the reported cyclo-(L-Leu-L-Leu-L-Leu-L-Leu-L-Ile).8 Five amide carbonyls and five α-methines were clearly observed. Thereafter, in order to confirm the structure, taking into consideration the importance of NH groups for assigning the amino acids sequence, we measured 1D and 2D NMR spectra of 1 in DMSO-d6 and found five down-field signals corresponding to amide protons (Table 1 and Fig. S5 and S6, ESI†). Furthermore, four Leu residues and one Ile were defined based on correlations from amide protons, α-proton, and butyl groups in 1H–1H COSY and TOCSY spectra (Fig. 3). Key HMBC correlations (Fig. 3) of the α-proton and/or NH of one amino acid residue with the carbonyl carbon of the neighboring residue were observed for determining the sequence of amino acids, which was also supported by the NOESY correlations (Fig. 3). Thus far, compound 1 was confirmed to be a pentapeptide, which should be cyclic.
:CD3OD (1:3) as the solvent for NMR measurements. The 1H and 13C NMR data (Fig. S3 and S4, ESI†) displayed the similar structural features as that for the reported cyclo-(L-Leu-L-Leu-L-Leu-L-Leu-L-Ile).8 Five amide carbonyls and five α-methines were clearly observed. Thereafter, in order to confirm the structure, taking into consideration the importance of NH groups for assigning the amino acids sequence, we measured 1D and 2D NMR spectra of 1 in DMSO-d6 and found five down-field signals corresponding to amide protons (Table 1 and Fig. S5 and S6, ESI†). Furthermore, four Leu residues and one Ile were defined based on correlations from amide protons, α-proton, and butyl groups in 1H–1H COSY and TOCSY spectra (Fig. 3). Key HMBC correlations (Fig. 3) of the α-proton and/or NH of one amino acid residue with the carbonyl carbon of the neighboring residue were observed for determining the sequence of amino acids, which was also supported by the NOESY correlations (Fig. 3). Thus far, compound 1 was confirmed to be a pentapeptide, which should be cyclic.
 | ||
| Fig. 1 The structures of compounds 1–4. | ||
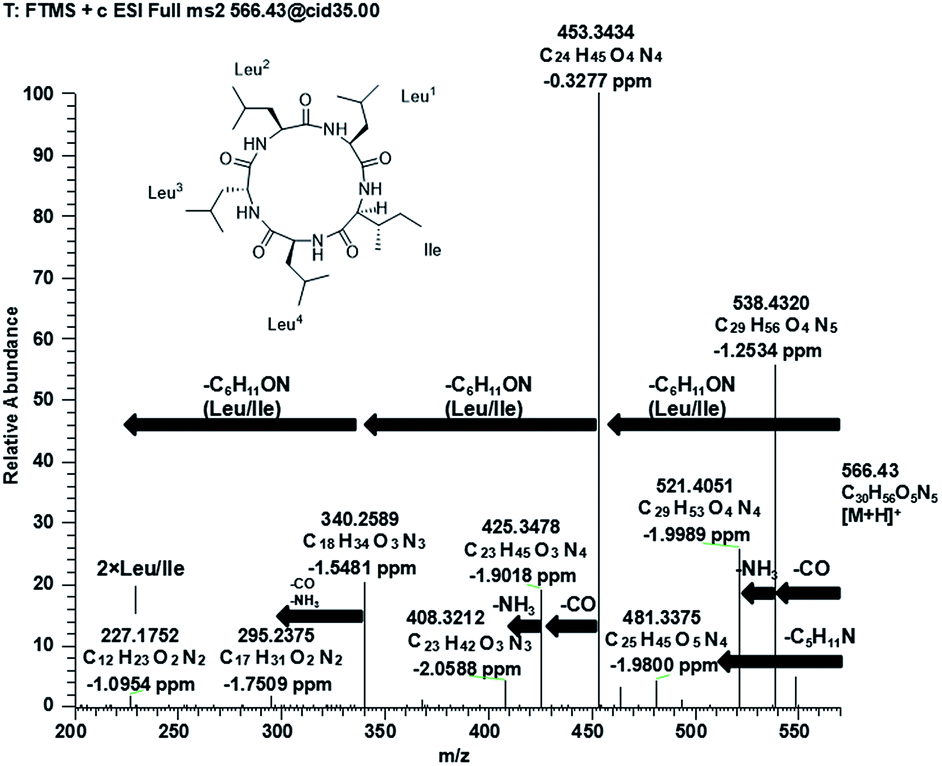 | ||
| Fig. 2 Positive MS/MS spectrum of compound 1. | ||
| Unit | No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|---|
| δ C, mult.a,d | δ H, mult.b (J in Hz) | δ C, mult.a,d | δ H, mult.b (J in Hz) | δ C, mult.a,d | δ H, mult.b (J in Hz) | ||
| a Recorded in DMSO-d6 at 125 MHz; 13C multiplicities were determined by HSQC experiment. b Recorded in DMSO-d6 at 500 MHz. c Signals overlapped. d Assigned by 2D NMR in DMSO-d6 together with the 13C NMR spectrum in CD3OD. | |||||||
| Leu1 | 1 | 171.3, Cq | 171.5, Cq | 171.5, Cq | |||
| 2 | 52.2, CH | 4.32, m | 52.2, CH | 4.32, m | 51.8, CH | 4.31, m | |
| 3 | 40.5, CH2 | 1.44, mc, 1.54, mc | 40.6, CH2 | 1.44, mc, 1.55, mc | 40.1, CH2 | 1.54, mc | |
| 4 | 24.8, CH | 1.44, mc | 25.1, CH | 1.44, mc | 25.0, CH | 1.47, mc | |
| 5 | 22.9, CH3 | 0.88, mc | 22.6, CH3 | 0.90, mc | 22.4, CH3 | 0.86, mc | |
| 6 | 22.7, CH3 | 0.90, mc | 23.1, CH3 | 0.86, mc | 23.1, CH3 | 0.90, mc | |
| NH | 7.20, d (6.5) | 7.21, br s | 7.27, br s | ||||
| Leu2 | 1 | 171.7, Cq | 172.0, Cq | 172.1, Cq | |||
| 2 | 52.5, CH | 4.07, m | 52.6, CH | 4.07, m | 52.4, CH | 4.10, m | |
| 3 | 40.2, CH2 | 1.50, mc | 40.4, CH2 | 1.51, mc | 40.4, CH2 | 1.50, mc | |
| 4 | 24.7, CH | 1.59, mc | 24.8, CH | 1.59, mc | 24.7, CH | 1.60, mc | |
| 5 | 21.0, CH3 | 0.77, d (6.0) | 21.1, CH3 | 0.78, d (6.5) | 21.1, CH3 | 0.78, d (6.5) | |
| 6 | 23.4, CH3 | 0.86, mc | 23.5, CH3 | 0.87, mc | 23.6, CH3 | 0.86, mc | |
| NH | 8.60, d (8.0) | 8.59, d (8.0) | 8.57, d (8.5) | ||||
| Leu3 | 1 | 172.0, Cq | 172.1, Cq | 172.1, Cq | |||
| 2 | 52.4, CH | 4.15, m | 52.5, CH | 4.17, m | 52.5, CH | 4.16, m | |
| 3 | 39.0, CH2 | 1.37, mc, 1.49, mc | 39.2, CH2 | 1.37, mc, 1.48, mc | 39.1, CH2 | 1.38, mc, 1.50, mc | |
| 4 | 25.1, CH | 1.49, mc | 24.8, CH | 1.45, mc | 24.8, CH | 1.46, mc | |
| 5 | 22.9, CH3 | 0.83, d (6.5) | 22.9, CH3 | 0.83, d (6.0) | 22.9, CH3 | 0.83, d (5.5) | |
| 6 | 22.5, CH3 | 0.88, mc | 22.5, CH3 | 0.88, mc | 22.5, CH3 | 0.88, mc | |
| NH | 8.73, d (6.0) | 8.71, d (6.5) | 8.74, d (6.5) | ||||
| Leu4 | 1 | 173.1, Cq | 173.3, Cq | 173.2, Cq | |||
| 2 | 50.6, CH | 4.44, m | 50.7, CH | 4.43, m | 50.8, CH | 4.38, m | |
| 3 | 42.1, CH2 | 1.36, mc | 42.2, CH2 | 1.37, mc | 42.3, CH2 | 1.37, mc | |
| 4 | 25.0, CH | 1.52, mc | 25.1, CH | 1.51, mc | 25.1, CH | 1.49, mc | |
| 5 | 23.6, CH3 | 0.88, mc | 23.3, CH3 | 0.88, mc | 23.3, CH3 | 0.88, mc | |
| 6 | 22.2, CH3 | 0.88, mc | 22.3, CH3 | 0.88, mc | 22.5, CH3 | 0.88, mc | |
| NH | 7.86, d (8.5) | 7.86, d (8.5) | 7.65, d (8.0) | ||||
| Ile (1) or Val (2) or Leu5 (3) | 1 | 171.6, Cq | 171.7, Cq | 172.8, Cq | |||
| 2 | 63.5, CH | 3.30, m | 65.6, CH | 3.20, t (8.5) | 56.3, CH | 3.77, m | |
| 3 | 33.6, CH | 2.30, m | 28.2, CH | 2.50, m | 39.0, CH2 | 1.54, mc, 1.82, m | |
| 4 | 25.7, CH2 | 1.03, m, 1.41, mc | 19.8, CH3 | 0.83, d (6.0) | 25.2, CH | 1.53, mc | |
| 5 | 10.3, CH3 | 0.79, t (6.5) | 20.1, CH3 | 0.83, d (6.0) | 22.0, CH3 | 0.83, d (5.5) | |
| 6 | 15.9, CH3 | 0.80, d (6.5) | 23.1, CH3 | 0.88, mc | |||
| NH | 8.38, br s | 8.42, br s | 8.21, d (6.0) | ||||
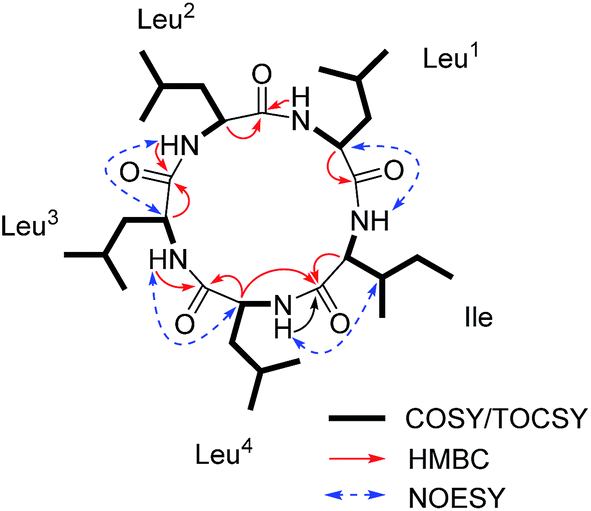 | ||
| Fig. 3 Key 1H–1H COSY/TOCSY, HMBC and NOESY correlations for 1. | ||
A single-crystal X-ray diffraction study with Cu Kα radiation allowed the assignment relative configuration of compound 1 as shown in Fig. 4. Finally, the absolute configuration of 1 was determined on the basis of the Flack parameter χ = 0.02(7). Interestingly, the crystal lattice displayed that three monomers were coupled together through five hydrogen bonds, affording a fascinating three-dimensional structure containing a “channel” inside (Fig. 4). The absolute configuration of five amino acid residues was also supported by applying the Marfey's method.9,10 The acid hydrolysate of 1 and the amino acid standards were derivatized with Marfey's reagent (FDAA) (Fig. S12, ESI†). LC-MS analysis (Fig. S13, ESI†) showed that the configuration of one leucine residue was D while that for others were L. The aforementioned analysis finally confirmed the structure of this pentapeptide as cyclo-(L-Leu-L-Leu-D-Leu-L-Leu-L-Ile).
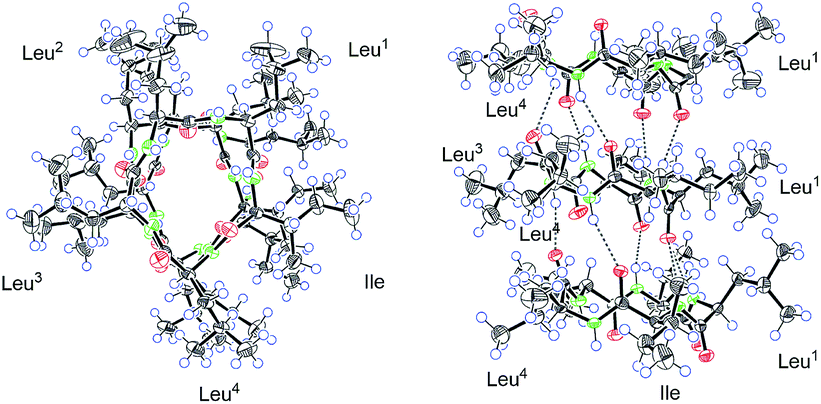 | ||
| Fig. 4 The Ortep drawing of compound 1 (left: a view from the top; right: a view from the side). | ||
The molecular formula of 2 (Fig. 1), C29H53O5N5 with six degrees of unsaturation, was assigned by ESI-HRMS at m/z 552.4124 [M + H]+ (calcd for C29H54O5N5, 552.4120, Δ 0.8478 ppm, Fig. S16, ESI†). Similar to compound 1, each of the five α-protons, α-methine carbons, and amide carbonyls were present in 1H and 13C NMR (in CD3OD) spectra (Fig. S18 and S19, ESI†). The high N content of 2 and its five NH signals ranging from δH 7.0 to 9.0 in 1H NMR spectrum (in DMSO-d6) also suggested it to be a pentapeptide (Table 1 and Fig. S20, ESI†). The interpretation of 1H–1H COSY and TOCSY data revealed the spin systems due to the amine protons (NH), α-protons, and additional high-field signals including methines, methylenes and methyl protons, verifying the presence of one valine (Val) and four Leu residues (Fig. 5). Moreover, these Val and Leu fragments were clearly observed in HR-CID-MS2 spectrum of 2 (Fig. S17, ESI†). Based on these results compound 2, a pentapeptide, should also be cyclic.
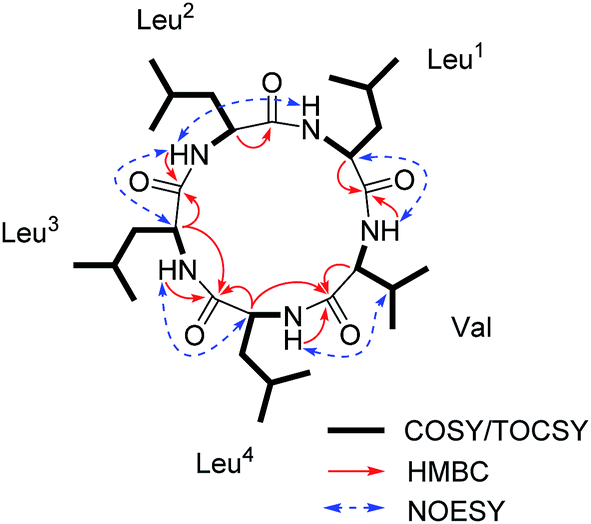 | ||
| Fig. 5 Key 1H–1H COSY/TOCSY, HMBC and NOESY correlations for 2. | ||
The sequence of the amino acids was confirmed by analysis of HMBC and NOESY data of 2 (Fig. 5). Key HMBC correlations of the α-proton and/or NH of one amino acid residue with the carbonyl carbon of the neighboring residue were observed from δH 8.59 (Leu2NH) to 172.1 (Leu3C-1), from δH 8.71 (Leu3NH) and δH 4.17 (Leu3H-2) to 173.3 (Leu4C-1), from δH 7.86 (Leu4NH) and δH 4.43 (Leu4H-2) to 171.7 (ValC-1), and from δH 8.42 (ValNH) to 171.5 (Leu1C-1). Cross-peaks of Leu1NH/Leu2NH, Leu2NH/Leu3H-2, Leu3NH/Leu4H-2, Leu4NH/ValH-3, and ValNH/Leu1H-2 in the NOESY spectrum of 2 finally determined the sequence as cyclo-(Leu1-Leu2-Leu3-Leu4-Val). The absolute configuration of five amino acid residues was established by applying the Marfey's method.9,10 The acid hydrolysate of 2 and amino acid standards were derivatized with Marfey's reagent (FDAA). LC-MS analysis showed that the configurations of one Val and three Leu residues were L (Fig. S27 and S28, ESI†). The remaining Leu unit was D-configured. It was located between Leu-2 and Leu-4, which was same as that of compound 1 and was supported by the key NOE cross peaks of Leu1NH/Leu2NH and Leu2NH/Leu3H-2 (Fig. 6). Consequently, the structure of 2 was determined as cyclo-(L-Leu-L-Leu-D-Leu-L-Leu-L-Val).
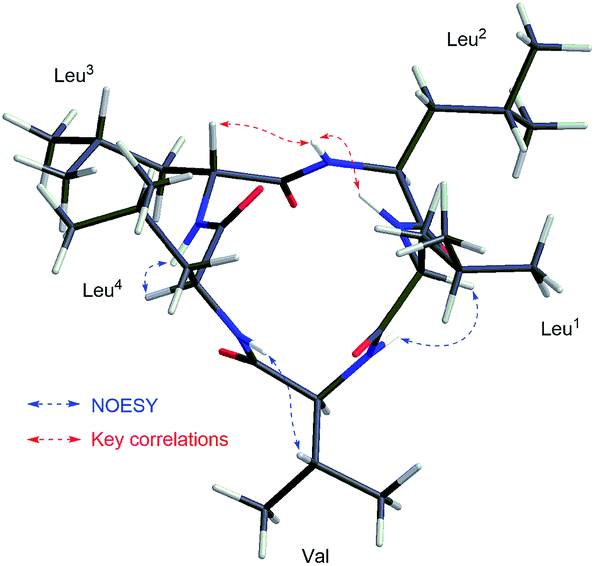 | ||
| Fig. 6 Key NOESY correlations shown in the 3D structure of compound 2. | ||
Compound 3 (Fig. 1) was obtained as a white powder, and its quasimolecular ion at m/z 566.4281 [M + H]+ (calcd for C30H56O5N5, 566.4276, Δ 0.8903 ppm; Fig. S31, ESI†) in ESI-HRMS spectrum indicated a molecular formula of C30H55O5N5, corresponding to the same molecular formula as 1 and with one CH2 group more than that of 2. Furthermore, HR-CID-MS2 spectrum (Fig. S32, ESI†) of 3, which showed the same fragment pattern as that of 1, displayed five Leu/Ile residues without a Val moiety. 1H and 13C NMR data (in CD3OD, Fig. S33 and S34, ESI†) of 3 revealed similar structural features as those of compounds 1 and 2. Further comparison of NMR data (in DMSO-d6) for 2 and 3 (Table 1) led to observation of significant differences in the downfield shift of H-2 (Leu5, δH 3.77 in 3; Val, δH 3.20 in 2) in 1H NMR spectrum of 3, as well as the upfield shift of C-2 (Leu5, δC 56.3 in 3; Val, δC 65.6 in 2), and an additional methylene carbon signal (Leu5, δC 39.0 in 3) in 13C NMR spectrum of 3. The above analysis indicated a pentapeptide made up of five Leu/Ile residues. By utilizing the same strategy as that for 1 and 2, a combination of COSY, TOCSY, HSQC, HMBC, and NOESY analysis (Fig. 7) along with the Marfey's method (Fig. S42 and S43, ESI†),9,10 the final structure of 3 was determined as cyclo-(L-Leu-L-Leu-D-Leu-L-Leu-L-Leu).
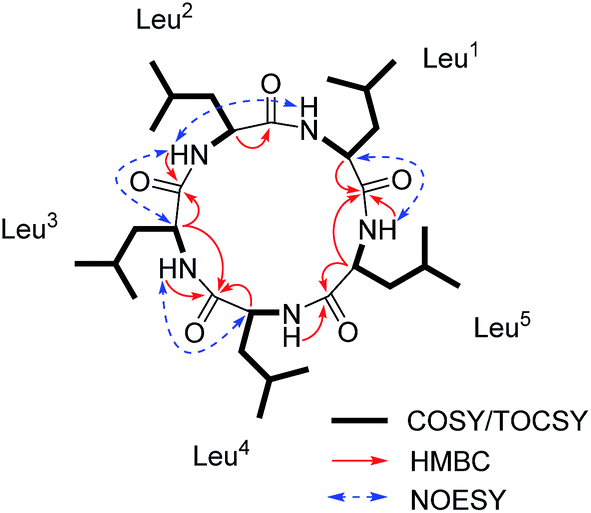 | ||
| Fig. 7 Key 1H–1H COSY/TOCSY, HMBC and NOESY correlations for 3. | ||
As expected from the above results, compounds 1–3 have a close similarity in their ECD spectra (Fig. S15, S30 and S45, ESI†) and optical rotation values. Compound 4 was identified to be fusaristatin A by the comparison of the MS and NMR data with that in literature (Fig. S49 and S50, ESI†).4 The structure of 4 was also strongly supported by the ESI-MS2 and MS3 spectra and the proposed fragmentation pathway (Fig. S47 and S48, ESI†).
The new compounds 1–3 were evaluated for in vitro antibacterial activities against five standard pathogenic control strains, namely Staphylococcus aureus (DSM 799), Escherichia coli (Risk Group 1; DSM 1116), E. coli (Risk Group 2; DSM 682), Bacillus subtilis (DSM 1088), and Acinetobacter sp. (DSM 586). Interestingly, none of them exhibited inhibitory activities against five tested strains. In literature,8 an analog of 1–3, cyclo-(L-Leu-L-Leu-L-Leu-L-Leu-L-Ile), exhibited only moderate inhibitory effects on three plant pathogenic fungi Aphanomyces cochlioides, Pythium ultimum and Rhizoctonia solani. Compound 4 has already been reported to have no antibacterial activities against S. aureus NBRC 13276, S. aureus ATCC 29213, S. aureus 25697, E. coli ATCC 25922, B. subtilis 168 trpC2, Pseudomonas aeruginosa ATCC 15442, P. aeruginosa B 63230, Streptococcus pneumoniae ATCC 49619, and Enterococcus faecalis UW 2689.4–6 It was also reported to be inactive against the fungi Candida albicans ATCC 2019, and Aspergillus clavataus F 318a.4
Given the weak antimicrobial activities of these peptides, along with our previous observation that certain cyclopeptides are used as chemical communication molecules by coexisting endophytes,7 we co-cultivated F. decemcellulare LG53 with another endophytic fungus G. acutata LG52 that was also isolated from the same stem tissue of the host plant (M. fortunei). The organisms were grown separately as well as in dual-culture setups for up to 7 days. As indicated in Fig. 8A and B, the fungus F. decemcellulare LG53 exhibited a slow-growing phenotype while the growth of G. acutata LG52 was much more pronounced. Interestingly however, when both fungi were growing together in a dual-culture, the growth of G. acutata LG52 was highly inhibited (Fig. 8C, 9A and C). This phenomenon led us to hypothesize that F. decemcellulare LG53 might be secreting one or more allelochemical(s) as a competitive strategy for survival in a limited ecological niche.11
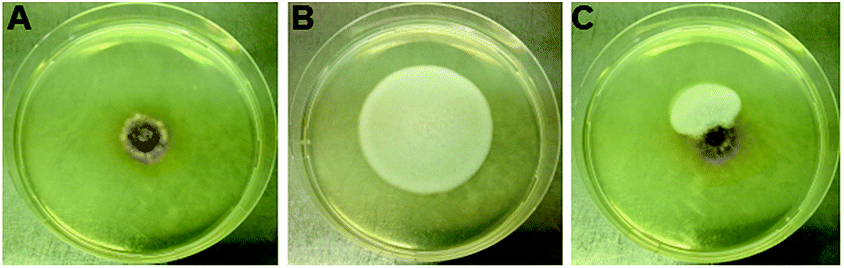 | ||
| Fig. 8 The co-culture between F. decemcellulare LG53 and G. acutata LG52 for 7 days. (A) The culture of fungus F. decemcellulare LG53 used as a control. (B) The culture of G. acutata LG52 used as a control. (C) Two fungi grown on PDA in a dual-culture format for 7 days. | ||
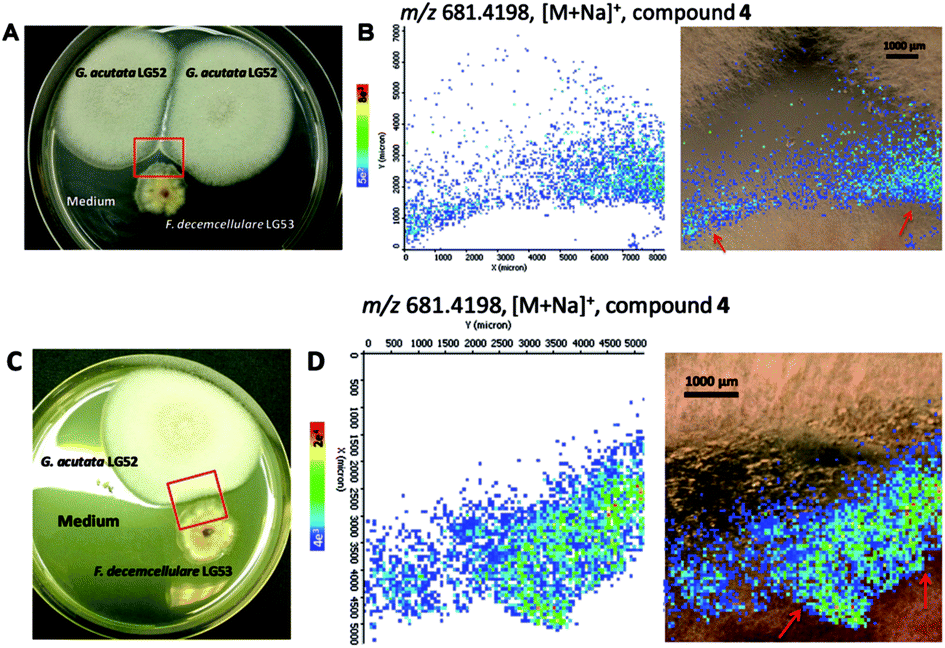 | ||
| Fig. 9 Accumulation of compound 4 produced by F. decemcellulare LG53 at the interface of the two endophytic fungi over 7 days, visualized by MALDI-imaging-HRMS experiments. (A and C) Optical image of co-culture between two endophytes and the area (red frame) selected for MS measurement. (B and D) Ion intensity maps of mass m/z 681.4198, C36H58O7N4Na, [M + Na]+ (compound 4). | ||
We tested the above hypothesis using two parallel approaches. Firstly, we used LC-HRMS to confirm the presence of compounds 1–4 as the main compounds produced by F. decemcellulare LG53 up to 7 days (Fig. S51, ESI†). In order to explore which main cyclic peptide(s) may be related to the inhibition of G. acutata LG52, we evaluated the antifungal activities of compounds 1–4 against G. acutata LG52 at different concentrations on PDA (as per our OSMAC results, 1–4 were the main compounds produced in this medium). Strikingly, the growth of G. acutata LG52 was inhibited by compound 4 (Fig. 10A), whereas compounds 1–3 did not inhibit the fungus. We noted that the inhibition of G. acutata LG52 by 4 was concentration-dependent (Fig. 10B). These results demonstrated that only compound 4 was active against the fungal endophyte G. acutata LG52, implying its possible role as an allelochemical leading to the observations in dual-cultures (Fig. 8C, 9A and C).
 | ||
| Fig. 10 Inhibition of G. acutata LG52 on PDA medium amended with compounds 1–4. (A) For each petri dish, there are total 400 μg pure compounds or mixture in 20 mL PDA medium (final concentration: 20 μg mL−1). (B) Compound 4 was amended into the PDA medium to reach the final desired concentrations before inoculating G. acutata LG52. | ||
In order to visualize the spatial distribution of metabolites particularly compound 4 in co-cultures, as our second approach, we employed MALDI-imaging-HRMS. It was observed in high spatial resolution that compound 4 was secreted into the PDA medium around the fungal mycelia of the producer F. decemcellulare LG53 (Fig. 9). Notably, compound 4 was accumulated outside F. decemcellulare LG53 with a higher ion intensity when the mycelia of the two endophytic fungi came in physical contact. These results cemented the fact that compound 4 is the allelochemical used by endophytic F. decemcellulare LG53 to ward off its associated endophyte G. acutata LG52. Moreover, the co-cultures of the fungi both on PDA as well as in liquid PDB media (7 days) were subjected to LC-MS analyses, indicating a higher ion intensity of compound 4 in co-cultures than that in the axenic culture of F. decemcellulare LG53 (Fig. S52 and S53, ESI†).
Taken together, our results reveal a proof-of-concept scenario of allelopathy (vis-à-vis balanced antagonism) wherein compound 4 could be used in situ by endophytic F. decemcellulare LG53 to inhibit coexisting microorganisms (such as G. acutata LG52). Notably, even though the fungal strain G. acutata LG52 exhibits an endophytic lifestyle in the Chinese medicinal plant M. fortunei, its morphologically distinct anamorph, Colletotrichum acutatum, has been shown not only to exhibit plant pathogenic lifestyle but also to possess entomopathogenic activity.12,13 Therefore, it is compelling that in the present study, endophytic F. decemcellulare LG53 employs compound 4 as an allelochemical against G. acutata LG52. This would provide an advantage to the producer in maintaining colonization of the host plant tissue as well as for warding off potential phytopathogens.1 Interestingly, compounds 1–3 were also secreted into medium (data not shown); however, they were not responsible in inhibiting G. acutata LG52.
The inimitable ecological function delivered by the endophytic F. decemcellulare LG53 lends further support to the multifarious lifestyle of Fusarium species ranging from saprophytic, pathogenic to endophytic. Notably, a plethora of plants residing in varied ecological niches harbor Fusarium species exhibiting an endophytic lifestyle and concomitantly, providing certain ecological fitness benefits. For instance, on the one hand, the plant Melia azedarach was shown to harbor different endophytic Fusarium species capable of producing fusariumin,14 fusarimine,15 fusaroside,16 fusarone,17 and other bioactive compounds unrelated to the host plant with plausible implications in aiding the host chemical defense. On the other hand, an endophytic strain of F. solani inhabiting the Chinese medicinal plant Camptotheca acuminata was shown to be capable of biosynthesizing the important anticancer compound camptothecin, also produced by the host plant, by means of a plant–endophyte cross-species biosynthetic pathway.18,19 More recently, another endophytic F. solani strain isolated from Narcissus tazetta plant of Chinese origin was shown to produce several hexacyclopeptides required to perform an important ecological function within the scope of microbial neighbor communication with a coexisting endophytic bacterium.7 These examples exemplify the complexities of plant–microbe and microbial interactions in different ecosystems, which play a major role in the production of secondary metabolites by endophytes and resulting in a variety of functional traits of both ecological and agricultural importance.
Experimental section
General experimental procedures
Optical rotations and IR spectra were measured through an A-Krüss Optronic P8000-T polarimeter and a Bruker Tensor 27 IR spectrometer, respectively. A Jasco J-715 spectrometer was employed to record ECD spectra. The NMR spectra were performed on a Bruker Avance DRX 500 spectrometer. LC-ESI-HRMSn data were measured on a LTQ-Orbitrap spectrometer (Thermo Fisher, USA) equipped with an Agilent 1200 HPLC system. Semi-preparative HPLC system included a Gynkotek pump, a Dionex DG-1210 degasser, a Dionex UVD 340S detector, a Dionex Gina 50 autosampler, together with a Venusil XBP C18 (2) column (10 × 250 mm, 5 μm). Silica gel 60 (70–230 mesh; AppliChem, GmbH, Darmstadt, Germany) and Sephadex LH-20 (25–100 μm; Amersham Biosciences) were applied as materials for column chromatography (CC). Thin-layer chromatography (TLC) was carried out with glass pre-coated silica gel 60 plates (0.25 mm; Merck, Darmstadt, Germany).Fungal material
The endophytic fungal strains were isolated from the stem of M. fortunei collected from Guangdong, People's Republic of China, following our previously established method.20 The endophytic fungus LG53 was identified as Fusarium decemcellulare by ITS sequencing (see detailed method in S55, ESI†).2 The endophytic fungus LG52 was identified as Glomerella acutata in a similar way. The ITS sequences have been deposited at the EMBL-Bank (awaiting accession numbers). Small-scale fermentations were performed using our previously established OSMAC approach.2 For large-scale fermentation, agar plugs from a week-old culture grown on PDA at 28 ± 2 °C were cut into small pieces and employed to inoculate 20 flasks (1 L) each containing 80 g of rice, 120 mL of water, and 0.3% peptone. The cultures were incubated at room temperature (RT) for 50 days.Extraction and isolation
The culture was extracted with EtOAc by sonication (20 min) at RT with the organic solvent completely submerging mycelia. The above step was repeated 3 times and the organic solutions were combined and evaporated under vacuum to dryness, affording 100 g crude extract. The extract was separated by CC (4.5 × 22 cm) on silica gel eluting with a gradient of cyclohexane and EtOAc (100:0–0:1, v/v) to yield 12 fractions (A–L). All the fractions were monitored by LC-HRMS to analyze their compositions. Fraction I (0.71 g) was subjected to the Sephadex LH-20 column (MeOH) to afford 7 subfractions (I1–I7). Subfraction I2 (207 mg) was purified by HPLC (MeOH–H2O, 85/15, 3.0 mL min−1) to obtain cyclo-(L-Leu-L-Leu-D-Leu-L-Leu-L-Ile) (1, 12.0 mg, tR = 22.4 min, purity > 98%), cyclo-(L-Leu-L-Leu-D-Leu-L-Leu-L-Val) (2, 4.5 mg, tR = 19.0 min, purity > 98%), and cyclo-(L-Leu-L-Leu-D-Leu-L-Leu-L-Leu) (3, 5.8 mg, tR = 20.3 min, purity > 95%). Fraction L (1.49 g) was also fractionated by Sephadex LH-20 column (MeOH) to afford 663 mg subtraction L1 (total 4 subfractions L1–L4), followed by HPLC purification (MeOH–H2O, 85/15, 2.5 mL min−1) to give fusaristatin A (4, 12.4 mg, tR = 23.8 min, purity > 98%).
:CD3OD 1:3, 500 MHz) and 13C NMR (CDCl3:CD3OD 1:3, 125 MHz), see ESI;† positive ESI-HRMS m/z: 566.4282 [M + H]+ (calcd for C30H56O5N5, 566.4276, Δ 1.1058 ppm).
X-ray crystallographic analysis of 1
The colorless crystals were obtained from a CH2Cl2:DMSO (1:1, v/v) solution. The single crystal X-ray diffraction analysis was carried out on a Bruker D8 Venture four-circle diffractometer equipped with PHOTON100 area Detector and Incoatec IμS radiation source with Helios mirror optics. The data was integrated with the Bruker SAINT software package using a narrow-frame algorithm. Data was collected at 100(2) K with Cu-Kα radiation (λ = 1.54178 Å). The multi-scan method (SADABS) was applied for data correction on absorption effects. The crystal structure was solved with intrinsic phasing using SHELXT2014 (ref. 21) and refined with the SHELXL2014 (ref. 22) refinement program by the full-matrix least-squares on F2 method. Molecular graphics were obtained using Ortep-3 (for Windows, Version 2014.1).23
Crystal data of 1: crystal size 0.060 × 0.108 × 0.153 mm3, triclinic crystal system, space group P1, unit cell dimensions a = 13.594(4) Å, b = 14.583(3) Å, c = 15.223(3) Å, volume 2477.2(10) Å3; Z = 3, T = 100(2) K, μ(Cu Kα) = 0.620 mm−1, Dcalc = 1.138 g cm−3; 62098 reflections collected with θ angle ranged from 3.35 to 67.99°; 17320 independent reflections (Rint = 0.0418). R1 = 0.0461, wR2 = 0.1187 (all data); absolute structure parameter (Flack parameter) 0.02(7).24 Crystallographic data of 1 was deposited in the Cambridge Crystallographic Data Centre with supplementary publication number CCDC 1476486.†
MALDI-imaging-HRMS
The methods for sample preparation(s) and measurement parameters were applied as described earlier.7Antimicrobial assays
The in vitro antibacterial activities of the compounds 1–3 were tested against a panel of standard pathogenic control strains (obtained from Leibniz-Institute DSMZ, Braunschweig, Germany) based on our previously described method.25 The antifungal activity of compounds 1–4 against the fungal strain G. acutata LG52 was evaluated by amending the compound(s) into the PDA media to reach the final desired concentrations. Agar plates were prepared in 90 mm sterile Petri dishes (TPP, Trasadingen, Switzerland) with 20 mL of PDA. The agar plates with the different concentrations of compound(s) were used to culture fungus. The agar plates without compound(s) were used as the control. Each test was carried out in triplicate.Acknowledgements
G. L. is grateful to the China Scholarship Council (CSC) for a doctoral fellowship. We thank the Ministry of Innovation, Science, Research and Technology of the State of North Rhine-Westphalia, Germany and the German Research Foundation (DFG) for funding a high-resolution mass spectrometer. We gratefully acknowledge Dr W. Hiller (Department of Chemistry and Chemical Biology, TU Dortmund) for the NMR measurements, Dr P. Kusari (formerly at the Department of Biochemical and Chemical Engineering, TU Dortmund) for technical assistance with identification of the fungal strain LG53, Mr S. Sezgin (INFU, TU Dortmund) for technical assistance with the MALDI-imaging-HRMS experiments, and Dr S. Zühlke (INFU, TU Dortmund) for realization of mass spectrometric analyses and valuable discussions.Notes and references
- S. Kusari, C. Hertweck and M. Spiteller, Chem. Biol., 2012, 19, 792–798 CrossRef CAS PubMed.
- G. Li, S. Kusari, P. Kusari, O. Kayser and M. Spiteller, J. Nat. Prod., 2015, 78, 2128–2132 CrossRef CAS PubMed.
- H. B. Bode, B. Bethe, R. Höfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS PubMed.
- Y. Shiono, M. Tsuchinari, K. Shimanuki, T. Miyajima, T. Murayama, T. Koseki, H. Laatsch, T. Funakoshi, K. Takanami and K. Suzuki, J. Antibiot., 2007, 60, 309–316 CrossRef CAS PubMed.
- A. R. B. Ola, D. Thomy, D. Lai, H. Brötz-Oesterhelt and P. Proksch, J. Nat. Prod., 2013, 76, 2094–2099 CrossRef CAS PubMed.
- A. Hegge, R. Lønborg, D. M. Nielsen and J. L. Sørensen, Metabolites, 2015, 5, 184–191 CrossRef CAS PubMed.
- W.-X. Wang, S. Kusari, S. Sezgin, M. Lamshöft, P. Kusari, O. Kayser and M. Spiteller, Appl. Microbiol. Biotechnol., 2015, 99, 7651–7662 CrossRef CAS PubMed.
- F. M. Talontsi, P. Facey, M. D. K. Tatong, M. T. Islam, H. Frauendorf, S. Draeger, A. von Tiedemann and H. Laatsch, Phytochemistry, 2012, 83, 87–94 CrossRef CAS PubMed.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- Y. Song, Q. Li, X. Liu, Y. Chen, Y. Zhang, A. Sun, W. Zhang, J. Zhang and J. Ju, J. Nat. Prod., 2014, 77, 1937–1941 CrossRef CAS PubMed.
- B. Schulz, S. Haas, C. Junker, N. Andrée and M. Schobert, Curr. Sci., 2015, 109, 39–45 Search PubMed.
- J. C. Guerber and J. C. Correll, Mycologia, 2001, 93, 216–229 CrossRef.
- J. Marcelino, R. Giordano, S. Gouli, V. Gouli, B. L. Parker, M. Skinner, D. TeBeest and R. Cesnik, Mycologia, 2008, 100, 353–374 CrossRef CAS PubMed.
- S.-X. Yang, J.-M. Gao, Q. Zhang and H. Laatsch, Bioorg. Med. Chem. Lett., 2011, 21, 1887–1889 CrossRef CAS PubMed.
- S.-X. Yang, J. Xiao, H. Laatsch, J. J. Holstein, B. Dittrich, Q. Zhang and J.-M. Gao, Tetrahedron Lett., 2012, 53, 6372–6375 CrossRef CAS.
- S.-X. Yang, H. P. Wang, J.-M. Gao, Q. Zhang, H. Laatsch and Y. Kuang, Org. Biomol. Chem., 2012, 10, 819–824 CAS.
- S.-X. Yang, J.-M. Gao, H. Laatsch, J. M. Tian and G. Pescitelli, Chirality, 2012, 24, 621–627 CrossRef CAS PubMed.
- S. Kusari, S. Zühlke and M. Spiteller, J. Nat. Prod., 2009, 72, 2–7 CrossRef CAS PubMed.
- S. Kusari, S. Zühlke and M. Spiteller, J. Nat. Prod., 2011, 74, 764–775 CrossRef CAS PubMed.
- S. Kusari, M. Lamshöft, S. Zühlke and M. Spiteller, J. Nat. Prod., 2008, 71, 159–162 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249–259 CAS.
- G. Li, S. Kusari, M. Lamshöft, A. Schüffler, H. Laatsch and M. Spiteller, J. Nat. Prod., 2014, 77, 2335–2341 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of compounds 1–4. CCDC 1476486. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra10905e |
| This journal is © The Royal Society of Chemistry 2016 |