
Affinity adsorbents for proline-rich peptide sequences: a new role for WW domains†
A. M. G. C. Diasa,
R. dos Santosa,
O. Iranzo*b and
A. C. A. Roque *a
*a
aUCIBIO, REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Campus Caparica, 2829-516 Caparica, Portugal. E-mail: cecilia.roque@fct.unl.pt; Fax: +351 21 294 8550
bAix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France
First published on 11th July 2016
Abstract
The WW domain derived from human Yes-associated protein (hYAP65_WW) recognizes proline-rich peptides. The structural and chemical robustness of WW domains makes them appealing candidates to target and capture these peptides in affinity purification processes. In this work, the chemical synthesis of the hYAP65_WW domain containing a terminal cysteine for oriented coupling onto the chromatographic matrix was successfully achieved by a fragment solution condensation reaction and by incorporation of pseudoproline dipeptide units. Both strategies yielded a hYAP65_WW protein with the characteristic WW domain folding. The purified hYAP65_WW domain was immobilized in a chromatographic matrix and tested for binding to a proline-rich peptide. The adsorbent bound 92 ng of peptide per mg of support and the elution was particularly efficient when employing a low pH or an increase in salt concentration. This work sets the ground for the application of WW domains as affinity reagents towards the capture and elution of peptides and proteins rich in proline sequences.
Introduction
Protein–protein interactions are critical for cellular regulation,1 as signal transduction pathways are mediated by the recognition of specific peptide sequences by proteins, in particular sequences rich in proline residues (Pro).Proline is a peculiar amino acid: the pyrrolidine ring confers rigidity to Pro-rich sequences and induces particular conformations. Despite its hydrophobic character, Pro also has an electron rich carbonyl group turning it into an exceptional hydrogen bond acceptor.2,3 Peptides with compositions of PXPXPXP or PPXPPXPP (X represents any amino acid), assemble into a so called PPI or a PPII helix turn formed by rotation of three residues, which forms a structural feature specifically recognized by different protein domains2 with affinity binding constants ranging from nM to μM.4 Pro-rich peptides and proteins are very frequent in humans,1 other animals (e.g. mouse), plants or bacteria (e.g. membrane proteins in Escherichia coli).5
The isolation and purification of Pro-rich peptides and proteins enables their further characterization and additional applications (e.g. study of molecular disease mechanisms6 and targets for drug design7). Purification of Pro-rich proteins and peptides is often cumbersome and based on affinity tag technologies or individually optimised protocols.8–10
Affinity chromatography represents an attractive strategy to isolate target molecules from a complex mixture, as this strategy can capture even very weak binders, which can be eluted and recovered for further identification and characterization.11
Peptides containing Pro-rich sequences are recognized by different families of domains including SH3 domain (Src-homology 3)12 and WW domains.13 WW domains possess 38–40 residues in length, with two tryptophan (W) residues spaced by 20–22 amino acids, which assemble on a three β-sheet structure.14 These motifs are present in 200 multidomain proteins and are usually localized in the recognition region, which is known to mediate protein–protein interactions.15 They have been classified in five different groups based on their recognition sequences. The human YAP65 WW domain (hYAP65_WW) is derived from the human Yes-associated protein, a proto-oncogene, and is representative of group I by recognizing PPX-Y, where X represents any amino acid.16,17 This domain has been associated to regulatory pathways important for cell growth and proliferation.18 It recognizes the peptide sequence EYPPYPPPPYPSG found in p53 (PY peptide, residues 742 to 754 of p53 binding protein-2; in bold are the main amino acids recognized by the WW domain).19 In this work the potential of hYAP65_WW to create affinity adsorbents for the recognition and capture of Pro-rich peptides, in particular those derived from p53, was assessed.
Results and discussion
In silico studies – stability and affinity
The hYAP65_WW domain possesses 44 residues in length (hYAP65_WWnativeFL). We shortened the sequence to focus on the amino acids crucial for recognition and folding, generating a smaller version of hYAP65_WW domain with 38 residues in length (hYAP65_WWnative). A Cys residue was also introduced at the N-terminal to facilitate coupling to the chromatographic support (hYAP65_WWmutated) (see Fig. 1A). | ||
| Fig. 1 Analysis of molecular dynamics results for three structures of hYAP65_WW domain. (A) Sequences used in the in silico studies derived from PDB databank code 1JMQ sequence, in the sequences K 30 was replaced by L; (B) RMSD (Root Mean Square Deviation) analysis; (C) RMSF (Root Mean Square Fluctuation) analysis; (D) B-factor analysis representation (figures produced using PyMol software). | ||
Molecular Dynamics (MD) simulations were employed to evaluate the stability of the hYAP65_WWmutated in comparison with the native structures. In Fig. 1B, the Root Mean Square Fluctuation (RMSD) shows that the three structures considered in this study become more stable after 25 ns, and this behaviour is maintained until the end of the simulation. In addition, the Root Mean Square Fluctuation (RMSF) (Fig. 1C) demonstrates that the structure hYAP65_WWmutated has lower flexibility in the overall structure compared with the other sequences, but maintains the high flexibility in loop I, known to be important for target recognition in WW domains. In all structures a higher flexibility was observed for the residues at the N and C terminals, with RSMF values of 0.6 to 0.8 nm as opposed to the other residues in the sequence showing RMSF values between 0.2 and 0.4 nm. This information can be visualized in the B-factor analysis (Fig. 1D), which indicates in a colour code the degree of flexible regions (dark blue – less flexibility, blue, green, yellow until red – high flexibility). In Fig. 1D, hYAP65_WWmutated shows a dark colour except in loop I and at the N and C terminals which appear in light blue colour. From the in silico studies it is also clear the formation of hydrophobic pockets between Trp39, Phe29, Pro42 and Trp19, Pro12, Pro14 and Pro42, which are important for the maintenance of the WW domain folding as described in the literature.19 During the time of the simulation the hydrogen bonds interactions were analysed and the most prevalent in all structures of hYAP65_WW are compared in Fig. S1A.† In Fig. S1B† it is possible to observe that the most prevalent hydrogen bonds correspond to interactions between the β-sheets and also in the C terminal region of the hYAP65_WW. Overall, hydrogen bonds and hydrophobic interactions contribute for the folding of the WW domain.
Molecular docking studies were then conducted to assess the interaction between hYAP65_WWmutated and a small version of the native peptide recognised by the hYAP65_WW domain. The peptide tested, PPPPYPAW, was derived from the p53 protein with the recognition sequence in bold, an Ala (spacer) and a Trp (fluorescent probe). The docking calculation yielded 41 clusters with estimated binding energy between −8.74 and −2.67 kcal mol−1. The best solution was presented in run 111 (78 conformations out of 256 runs) with an estimated KD of 3.62 × 10−7 M (Fig. 2A). This value is lower than the literature reports for the full length peptide where a KD of 5.99 × 10−6 M was experimentally observed.19 In this docking solution, the peptide is recognized by a hydrophobic cluster formed by several amino acids from the β-sheets of hYAP65_WWmutated. The Tyr in the peptide PPPPYPAW is recognized through a groove formed by Tyr28 and Trp39, as previously described by Koepf et al.19 In this conformation the hydrophobic interactions between the Tyr and Trp28 and also the fluorescent probe with Tyr28 could be relevant, as the amino acids are in close proximity (∼4 Å). In the second best docking conformation, run 142 with KD of 2.18 × 10−7 M, the Tyr of the peptide is close to His32 with possible formation of hydrogen bonds (Fig. 2B). This is an important interaction already described in the literature for recognition between PPXY peptides and WW domains.13
 | ||
| Fig. 2 Best docking results for hYAP65_WWmutated and PPPPYPAW peptide. (A) Cluster with 78 similar conformations out of 256 possible conformations, estimated binding energy of −8.74 kcal mol−1. The interactions between the peptide Tyr or Trp and the Tyr28 and Trp39 are represented; (B) cluster with 43 similar out of 256 possible conformations, estimated binding energy of −6.36 kcal mol−1. The interaction between the peptide Tyr and the His32 of the hYAP65_WWmutated is represented (2.2 Å distance between Tyr OH-group and NH-group in the imidazole ring of His32). Structures produced using PyMol software. | ||
Solid-phase peptide synthesis
The hYAP65_WWmutated peptide was produced chemically using Fmoc-based chemistry and an automated microwave peptide synthesizer. A standard protocol for synthesis of hYAP65_WW domain was tested. The crude peptide obtained was analyzed by reverse-phase HPLC and the chromatogram showed several major peaks (Fig. S2†) indicating a very complex mixture that will be very difficult to purify. For this reason, new strategies were needed to improve the synthesis. WW domains possess a characteristic secondary structure composed by three β-sheets which increases the tendency for aggregation during synthesis, thereby reducing the coupling efficiency and yielding complex mixtures hard to purify. To overcome this problem, several strategies are presented in the literature: namely the use of different solid-supports, e.g. poly(ethylene glycol) (PEG) based resins;20 the change of solvents;21 the incorporation of special amino acid units during synthesis, e.g. pseudoproline dipeptide units, depsipeptides;22 or two or more fragments condensation, using for example native chemical ligation.23The hYAP65_WWmutated peptide was produced through two different strategies: solution condensation reaction of two fragments (SCR) and incorporation of pseudoproline dipeptide units during full peptide synthesis (PP) (Fig. 3). For the SCR strategy, two different resins were selected to synthesize the protected fragments (Frag1 in amide Sieber resin (17 residues) and Frag2 in H-Gly-2-Cl-trityl resin (21 residues)) with the required free N and C terminals for the subsequent condensation reaction (Fig. 3A, see reaction details in Experimental section). After deprotection, the crude hYAP65_WWmutated_SCR peptide was purified by preparative HPLC and characterized by ESI-MS (Fig. 3E). In the PP synthesis protocol, a PEG-based resin and two pseudoproline units were used (see Fig. 3B for their location), following a recently described method.24 The peptide hYAP65_WWmutated_PP was also purified by preparative HPLC and characterized by ESI-MS (Fig. 3F).
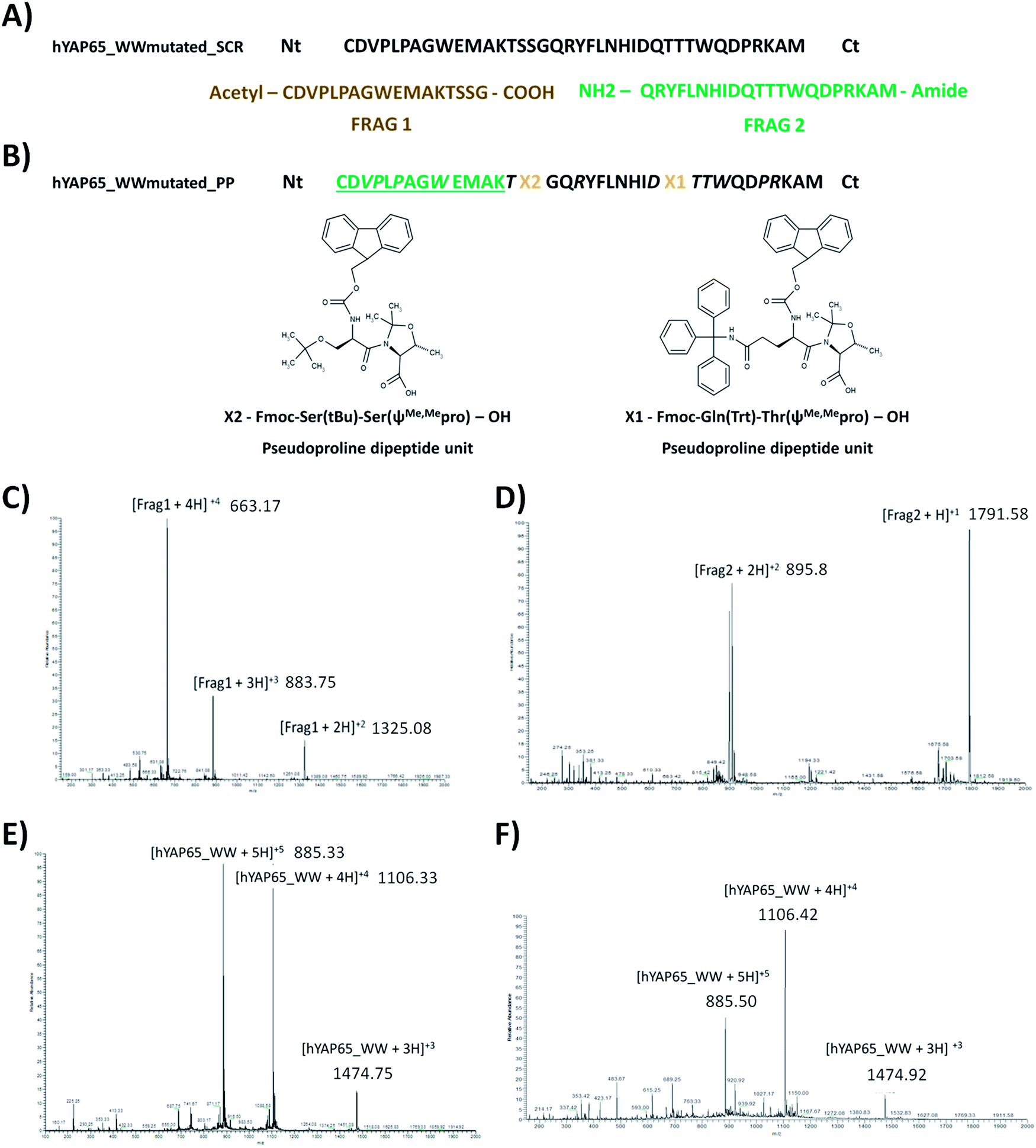 | ||
| Fig. 3 Nomenclature and amino acid sequences for different hYAP65_WW domain used in this work. (A) Solution Condensation Reaction (SCR); (B) synthesis using pseudoprolines (PP), where X1 and X2 are pseudoproline units; amino acids in italic needed special couplings and for amino acids in green and underlined, a capping step was added. The structures were designed using Marvin Beans version 6.3.0 (ChemAxon). Peptides were purified using preparative HPLC, characterized by ESI-MS in positive mode (for details see ref. 24). (C) Frag1-SCR: [Frag1 + 1]calc+1 = 2649 Da. (D) Frag2-SCR: [Frag2 + 1H]calc+1 = 1791 Da. (E) hYAP65_WWmutated_SCR peptide: [YAP65 + 1H]calc+1 = 4421 Da. (F) hYAP65_WWmutated_PP peptide: [YAP65 + 1H]calc+1 = 4421 Da. | ||
Circular dichroism studies
Circular Dichroism (CD) spectroscopic studies were performed to determine the structure and stability of the chemically synthesised hYAP65_WWmutated_SCR and hYAP65_WWmutated_PP. The spectra were recorded in the far-UV region to check for the characteristic signals of the WW domains. These proteins have a three β-sheet structure showing a maximum positive ellipticity at 230 nm and a maximum negative ellipticity at 206 nm in the CD spectra (Fig. 4). Fig. 4A and C show the CD spectra obtained for hYAP65_WWmutated_SCR and hYAP65_WWmutated_PP, respectively, at 4 °C and pH 6. In both cases, a maximum positive ellipticity at 230 nm was observed, as expected for a WW domain.19 In addition, temperature denaturation experiments were performed to determine the stability of the peptides. The change in ellipticity at 230 nm was monitored as the temperature increased from 4 °C to 88 °C and vice versa (Fig. S3A and B†). The CD melting curves were fitted to a two state-model, using the equation described by Koepf et al.19 to determine the temperature of melting (Tm) of the two hYAP65_WW. | ||
| Fig. 4 Circular dichroism studies. (A) Far-UV CD spectra at 4 and 88 °C, hYAP65_WWmutated_SCR; (B) unfolding and fitting curve between 4 and 88 °C, hYAP65_WWmutated_SCR; (C) far-UV CD spectra at 4 and 88 °C, hYAP65_WWmutated_PP; (D) unfolding and fitting curve between 4 and 88 °C, hYAP65_WWmutated_PP. | ||
The Tm determined by Koepf et al. for the peptide hYAP65_WW (57 aa) was 48.9 ± 0.6 °C.19 In our studies the Tm determined was 31.42 ± 2.95 °C (R2 = 0.996) and 28.52 ± 2.66 °C, (R2 = 0.993) for hYAP65_WWmutated_SCR and hYAP65_WWmutated_PP, respectively (Fig. 4B and D, S3A and B†). These data indicate a loss of thermal stability in the smaller versions of the hYAP65 (38 aa) with respect to the full-length hYAP65_WW domain (57 aa). Nonetheless, the CD spectra show that hYAP65_WWmutated_SCR and hYAP65_WWmutated_PP structures still maintained the characteristic folding of a WW domain and the unfolding state is totally reversible for both peptides.
Immobilization of hYAP65_WWmutated and affinity studies
As the hYAP65_WW domain recognizes Pro-rich peptides we aimed to test if the smaller WW domain versions chemically produced, still maintained the recognition ability after covalent immobilization on a matrix. For that purpose, we immobilized hYAP65_WWmutated_PP onto cross-linked agarose beads using Sulfo-SMCC chemistry (54% immobilization yield; 2.42 × 10−3 μmol hYAP65_WWmutated per mg of support). The novel affinity adsorbent (hYAP65_WWAg) was tested for binding to a Pro-rich peptide (PPPPYPAW). As these assays were performed at 23 °C, we considered that 30% of the peptide was folded, as described in the Experimental section. The hYAP65_WWAg bound 92.1 ± 20.9 ng of peptide per mg support (Table 1), which is 2.4 times higher than the negative control (unmodified agarose). Two peptides without Pro-rich sequences and with distinct hydrophilicity and charge – NNNNNN and RKRKRK – were tested as controls for binding to the modified agarose. No binding was observed for these peptides.| a Peptide bound mass/support mass (ng mg−1) = (peptide mass loaded − peptide mass unbound)/support mass.b % recovery = (peptide mass eluted × 100)/peptide mass bound. | |||||
|---|---|---|---|---|---|
| Binding | ng peptide bound per mg supporta (n = 12) | 92.1 ± 20.9 | |||
| Elution | 0.5 M NaCl | 1 M NaCl | pH 3 | pH 10 | |
| % recoveryb (n = 2) | 7.4 ± 1.5 | 55.1 ± 7.9 | 68.2 ± 1.6 | 34.2 ± 0.6 | |
Two different elution conditions were then tested to recover the Pro-rich peptide bound (Table 1): (a) an increase in ionic strength, by a step change of NaCl concentration from 0.5 M to 1 M and (b) a change in pH buffer, by using a pH 3 buffer (10 mM glycine–HCl) followed by a pH 10 buffer (10 mM CAPS buffer, 100 mM NaCl). The recovery of the peptide was more efficient when employing a decrease in pH (pH 6 to pH 3), with 68% recovery yield. This can be explained by the interactions established between the OH-group of the Tyr in the peptide and the NH-group in the imidazole ring of His32 (from WW domain) (an interaction with a distance of 2.2 Å, Fig. 2B). At pH 3 the imidazole NH-group has a positive charge (pKa = 6) which weakens the interaction with the OH group of Tyr, thus facilitating the elution of the peptide. The elution observed at a low pH can also be due to a decrease in WW folding – it was recently reported that at pH 3 the WW stability is decreased, yet this behaviour is reversible.25 At pH 10, the OH-group from the Tyr residue is deprotonated and there is no possibility to form hydrogen bonds with the imidazole N from the imidazole ring of His 32. Consequently, this pH condition is also a viable condition for elution. Therefore, extremes of pH (3 or >10) would be good to be used for elution, due to the disruption of the His32 interaction with Tyr of PPPPYPAW.
Recovery using a salt gradient was also possible, in particular when employing a high salt concentration solution (1 M NaCl). By increasing the salt concentration the hydrophobic effect is increased as the salt in the buffer reduces the solvation of sample solutes and hydrophobic regions become more exposed. This will affect the disposition of the residues and the distances between them. The recognition between the WW domain and the peptide is mediated by hydrophobic interactions in the “X–P groove” (formed by Trp and Tyr) and the Pro residues in the peptide, which is maintained by a tight control of atom distances.2 Therefore, if we interfere with these distances the hydrophobic interactions decrease and elution can occur.
Static partitioning equilibrium studies adjusted to a Hills isotherm model were employed to assess the values of qmax (1471 ± 462 ng peptide bound per mg support) and the dissociation constant (KD = 1.3 × 10−4 M) (see Fig. 5 and Table 2). The KD value between the peptide and immobilised WW is higher than that described in the literature namely for the interaction between the peptide EYPPYPPPPYPSG and WW free in solution (KD = 5.99 × 10−6 M).19 The difference in the results can be due to the immobilization of the WW domain in the agarose matrix. It is known that the spacer arm between the affinity ligand and the resin, the chemistry employed for the immobilization, and the nature of the solid support, can all have an effect on the dissociation constant observed in solid-phase. Another aspect that can contribute for the observed differences is the fact that the target peptide used in this work is smaller than the one described by Koepf et al. As previously described by Dalby et al.26 the difference in binding capacity can be influenced by the residues that are at the edges of the region that is being recognized, in this case the PPPPYP. Still, a KD in the range of 10−4 M is desirable in affinity chromatography applications as it facilitates the recovery of the target peptides and proteins.
 | ||
| Fig. 5 Characterization of binding between hYAP65_WWAg and peptide PPPPYPAW. Solid line represents the fit to the Hills equation with a n = 1, R2 = 0.999. | ||
| Binding characterization | qmax (ng peptide bound per mg support) | 1471 ± 462 |
| KD (M) | 1.3 × 10−4 ± 7.9 × 10−5 |
Conclusions
In this work we demonstrated the application of hYAP65_WW domain as a novel affinity ligand for the purification of Pro-rich peptides. The hYAP65_WW is an interesting affinity protein due to its accessible chemical synthesis. This protein can be used for the purification of Pro-rich peptides using mild conditions for binding and a pH variation for elution. Thus this domain can be explored for biotechnology applications, namely affinity purification for the enrichment of proline rich peptides and its further integration in proteomic studies.Experimental
In silico studies
Reagents
N,N-Dimethylformamide (DMF), acetonitrile, dichloromethane (DCM) and N-methylpyrrolidone (NMP) were purchased from Fisher Scientific (Loures, Portugal), N,N-diisopropylethylamine (DIEA), piperidine, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), trifluoroacetic acid (TFA), thioanisole, 1,2-ethanedithiol, anisole, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS), 1,4-diaminobutane and sodium chloride (NaCl) were purchased from Sigma-Aldrich (Loures, Portugal). Glycine > 99.5% was purchase from NZYtech (Lisbon, Portugal). All Fmoc protected amino acids, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) resins and pseudoproline units, were purchased from EMD Biosciences/Merck Biosciences (Darmstadt, Germany). Sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC) and bond-breaker TCEP solution purchased were from ThermoScientific, Portugal. Sepharose CL-6B was purchased from GE Healthcare Life Sciences, Portugal. The reagents used in these procedures were always high grade.Peptide chemical synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5), 2 h; Frag2: TFA/thioanisole/1,2-ethanedithiol/anisole (% v/v = 90:5:3:2), 2 h). The crude peptides were analysed by reverse-phase preparative HPLC, using the following conditions: Phenomenex Jupiter Proteo column (250 mm × 21.20 mm, 4 μ, 90 Å) and a linear gradient from 20% to 50% B in 25 min (solvent A (water/TFA, 99.9:0.1 v/v) and solvent B (acetonitrile/water/TFA, 90:9.9:0.1 v/v)). In each case, a major peak was detected which was collected and analyzed by ESI-Mass Spectrometry (ESI-MS). The ESI-MS spectra recorded in the positive mode confirmed the identity of the two fully protected fragments. Subsequently, both protected fragments were cleaved from the acid-labile linker resins. For the 2-Cl-trityl resin, short treatments of 2 min with 1% TFA/DCM were carried out. For the Sieber amide resin, longer treatments (5 min) and higher content of TFA (2.5% TFA/DCM) were required. The solutions were filtered into a vial containing DIEA to neutralize the pH and those fractions that contained the peptides, as shown by TLC, were combined and the solvent reduced under vacuum. Small volumes of water were used to precipitate the protected peptide. The precipitate was dried using the vacuum pump. Both fragments were used without further purification.
:2.5:2.5), 2 h; Frag2: TFA/thioanisole/1,2-ethanedithiol/anisole (% v/v = 90:5:3:2), 2 h). The crude peptides were analysed by reverse-phase preparative HPLC, using the following conditions: Phenomenex Jupiter Proteo column (250 mm × 21.20 mm, 4 μ, 90 Å) and a linear gradient from 20% to 50% B in 25 min (solvent A (water/TFA, 99.9:0.1 v/v) and solvent B (acetonitrile/water/TFA, 90:9.9:0.1 v/v)). In each case, a major peak was detected which was collected and analyzed by ESI-Mass Spectrometry (ESI-MS). The ESI-MS spectra recorded in the positive mode confirmed the identity of the two fully protected fragments. Subsequently, both protected fragments were cleaved from the acid-labile linker resins. For the 2-Cl-trityl resin, short treatments of 2 min with 1% TFA/DCM were carried out. For the Sieber amide resin, longer treatments (5 min) and higher content of TFA (2.5% TFA/DCM) were required. The solutions were filtered into a vial containing DIEA to neutralize the pH and those fractions that contained the peptides, as shown by TLC, were combined and the solvent reduced under vacuum. Small volumes of water were used to precipitate the protected peptide. The precipitate was dried using the vacuum pump. Both fragments were used without further purification.
| Reagents | Power (watts) | T (°C) | Reaction time (s) | |
|---|---|---|---|---|
| a Arginine residues requires double coupling: 1st coupling.b Arginine residues requires double coupling: 2nd coupling. | ||||
| Deprotection | 20% piperidine/DMF | 35 | 50 | 234 |
| Coupling | HBTU/DIEA/DMF | 25 | 50 | 390 |
| Coupling Arg (double coupling) | HBTU/DIEA/DMF | 0a and 25b | 50 | 1950a and 400b |
The condensation reaction was carried out using the following procedure. A reaction mixture containing 1.2 eq. Frag2, 1.2 eq. PyBOP and 4 eq. DIEA in DCM was stirred for 5 min under nitrogen. Afterwards, 1.2 eq. of Frag1 were added. To increase the solubility, the protected Frag1 and Frag2 were dissolved in a minimum amount of DMF before addition into the DCM mixture. After 1 h reaction, extra 1.2 eq. of PyBOP and 4 eq. of DIEA were added. The reaction was monitored by analytical reverse-phase HPLC by taking aliquots during the reaction period. The following HPLC conditions were used: Phenomenex Jupiter Proteo column (250 mm × 4.60 mm, 4 μ, 90 Å) a 30% solvent B isocratic flow for 5 min follow by a 10 min linear gradient until 40% solvent B (solvent A (water/TFA, 99.95:0.05 v/v) and solvent B (acetonitrile/water/TFA, 90.95:9:0.05 v/v)), with 0.6 mL min−1 flow rate. Rt (Frag1-protected) = 3.76 min, Rt (Frag2-protected) = 6.10 min and Rt (product-protected) = 6 min. The reaction took place for 10 h at room temperature under nitrogen atmosphere and magnetic stirring. This reaction time was chosen since similar strategies described in the literature demonstrated that increasing reaction time to 24 h did not improve the reaction yield.32 Indeed, in our case, longer reaction times produced a mixture of compounds as observed by HPLC analysis (results not shown). At the end of the reaction, solvents were removed and a yellow oil was obtained, which yielded a white precipitate (protected peptide) upon addition of water. After filtration and drying, the full deprotection reaction was carried out using TFA/thioanisole/1,2-ethanedithiol/anisole (% v/v = 90:5:3:2) for 2 h at room temperature and under nitrogen atmosphere. The resultant crude peptide was purified by reverse-phase HPLC using the following conditions: Phenomenex Jupiter Proteo column, 250 mm × 21.2 mm, 4 μ, 90 Å, linear gradient from 20% to 50% solvent B over 25 min with a flow rate of 10 mL min−1, solvent A (water/TFA, 99.9:0.1 v/v) and solvent B (acetonitrile/water/TFA, 90:9.9:0.1 v/v). The pure peptide was characterized by ESI-MS. The purity of the peptide (hYAP65_WWmutated_SCR) was checked by analytical reverse phase HPLC and it was greater than 95%.
Pseudoproline units were used to simplify the synthesis protocol by preparing a full length hYAP65_WWmutated peptide. This strategy was applied previously for the synthesis of human Pin1 WW domain.24 This strategy was feasible because this domain contains Ser and Thr residues located in the middle of the sequence. Consequently, two dipeptide units, Fmoc-Gln(Trt)-Thr(ψMe,Mepro)-OH and Fmoc-Ser(tBu)-Ser(ψMe,Mepro)-OH were selected for introduction during synthesis (see Fig. 3B for specific location). The synthesis (0.25 mmol scale) was carried out using a NovaPEG Rink Amide LL resin (substitution 0.23 mmol g−1, Novabiochem), which was previously swelled for 30 min in DMF and directly loaded in the reaction vessel. The protocol for couplings and reaction mixtures was the same as described before.24 After synthesis the resin was treated with the mixture TFA/thioanisole/1,2-ethanedithiol/anisole (% v/v = 90:5:3:2) for 3 hours at room temperature and under nitrogen to cleave and fully deprotect the peptide. The solution was reduced under a nitrogen stream and cold diethyl ether was added to precipitate the crude peptide which was dissolved in water and lyophilized. hYAP65_WWmutated_PP crude sample was purified by preparative reverse-phase HPLC, using the solvent B mixture described before. The peptide was eluted with a linear gradient of solvent B (20–50% in 25 min) at a flow rate of 10 mL min−1 (Rt = 20.52 min). The peptide was characterized by ESI-MS and the molecular weight was determined.
Immobilization of hYAP65_WWmutated and affinity studies
To proceed with immobilization of hYAP65_WWmutated in the aminated support, the following solutions were used: an immobilization buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA pH 7.2) and a buffer to dissolve Sulfo-SMCC (50 mM HEPES pH 7). These buffers were de-aerated using nitrogen flow before use. Sulfo-SMCC was used in a 5 molar excess to the number of amines in the support, this is in accordance with manufacturer indications, to achieve a ratio of 0.5 μmol peptide: 1 μmol of aminated support. The support was washed 5 times with 500 μL of immobilization buffer. The solution of 9.5 mg Sulfo-SMCC in 950 μL of 50 mM HEPES pH 7 was prepared, added to the support and incubated with orbital agitation (30 rpm) for 30 min at room temperature. Meanwhile, a solution of hYAP65_WWmutated (10 mg mL−1) was prepared in immobilization buffer and sonicated for 10 min. To avoid any oxidized Cys in the peptide, bond-breaker TCEP solution reagent was added in a volume ratio of 1:100 peptide/TCEP. After incubation the solution was removed by filtration and the support was washed 3 times with deaerated immobilization buffer to eliminate unreacted Sulfo-SMCC. After this, the solution of hYAP65_WWmutated was added to the modified support and incubated for 1 h in the same conditions described above. The support was washed with immobilization buffer 4 times and a solution of L-cystein was prepared in the same molar ratio of Sulfo-SMCC and incubated for 30 min with the support to block non-reacted sites. Afterwards, the support was washed 4 times with immobilization buffer. Finally, to determine the yield of immobilization, all samples obtained during the washing procedure were quantified by intensity fluorescence (λexcitation = 280 nm and λemission = 340 nm, calibration curve: y = 397501x, R2 = 0.99 and a gain: 57) using a microplate reader Tecan instrument. The support (hYAP65_WWAg) was saved at 4 °C in immobilization buffer.
000 rpm for 2 min. The binding buffer used in these studies was 10 mM sodium phosphate, 100 mM NaCl pH 6, the same buffer used by Koepf et al.19 in binding studies with native ligand of hYAP65_WW. The support was equilibrated with binding buffer, until the absorbance of the samples reached Abs280 nm < 0.005. The peptide PPPPYPAW (purity > 97% from CASLO, ApS, Lyngby, Denmark) was reconstituted at 0.20 mg mL−1 in binding buffer. This solution (0.30 mL) was incubated at room temperature (23 °C) for 2 h with the equilibrated hYAP65_WWAg adsorbent. After incubation, the flow-through was collected and the support was washed 7 times with 0.30 mL binding buffer. As a negative control, unmodified agarose was also tested in the same conditions. The amount of peptide in all collected samples, flow-through and washes was analyzed by measuring the Abs230 nm and intensity fluorescence (λexcitation = 280 nm and λemission = 340 nm) in a microplate reader Tecan instrument to determine the amount of PPPPYPAW bound to the support. The same conditions were used to test two peptides without Pro-rich sequences – NNNNNN and RKRKRK (purity > 97% from CASLO, ApS, Lyngby, Denmark). The binding results were treated to determine the amount of peptide bound to the support and expressed as ng peptide bound per mg support = (ng peptide loaded − ng peptide output) per mg support, the amount of support was multiplied by 30% to account for the amount of folded peptide at 23 °C. The elution conditions tested in independent experiments were salt and pH step gradients: (i) 25 mM HEPES buffer pH 7 with 0.5 M NaCl followed by 25 mM HEPES buffer pH 7 with 1 M NaCl; (ii) 10 mM glycine–HCl pH 3 followed by 10 mM CAPS buffer, 100 mM pH 10. The supports were washed with each elution buffer 4 times 0.15 mL. The amount of peptide in the elution samples was determined by measuring intensity fluorescence (λexcitation = 280 nm and λemission = 340 nm) in microplate reader Tecan instrument. The elution results were expressed as the amount of ng peptide eluted per mg support, the amount of support was multiplied by 30% to account for the amount of folded peptide at 23 °C and % recovery = (peptide mass eluted × 100)/peptide mass bound.
 | (1.1) |
Acknowledgements
A. M. G. C. D. would like to thank Dr Iris Batalha and Dr Ricardo Branco for the helpful discussions and technical support in peptide immobilization and in the in silico studies, respectively. A. M. G. C. D. and R. S. are grateful to Fundação para a Ciência e Tecnologia (FCT) – Portugal for funding through PhD grants SFRH/BD/72664/2010 and PD/BD/105753/2014, the National Network of Mass Spectrometry REDE/1504/REM/2005, project PTDC/EBB-BIO/102163/2008 and PTDC/EBB-BIO/118317/2010. This work was supported by the Unidade de Ciências Biomoleculares Aplicadas-UCIBIO which is financed by national funds from FCT/MEC (UID/Multi/04378/2013) and co-financed by the ERDF under the PT2020 Partnership Agreement (POCI-01-0145-FEDER-007728). Mass Spectrometry data were provided by the Mass Spectrometry Laboratory, Analytical Services Unit, Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa.References
- A. A. Morgan and E. Rubenstein, PLoS One, 2013, 8, e53785 CAS.
- L. J. Ball, R. Kühne, J. Schneider-Mergener and H. Oschkinat, Angew. Chem., 2005, 44, 2852–2869 CrossRef CAS PubMed.
- W. Gu and V. Helms, Biochim. Biophys. Acta, 2005, 1754, 232–238 CrossRef CAS PubMed.
- J. A. Jadwin, M. Ogiue-Ikeda and K. Machida, FEBS Lett., 2012, 586, 2586–2596 CrossRef CAS PubMed.
- M. P. Williamson, Biochem. J., 1994, 297, 249–260 CrossRef CAS PubMed.
- J. H. Cho, C. B. Park, Y. G. Yoon and S. C. Kim, Biochim. Biophys. Acta, 1998, 1408, 67–76 CrossRef CAS.
- M. Srinivasan and A. K. Dunker, Int. J. Pept., 2012, 2012, 1–14 CrossRef PubMed.
- G. S. Bedi and S. K. Bedi, Prep. Biochem., 1995, 25, 119–132 CrossRef CAS.
- H. Boze, T. Marlin, D. Durand, J. Pérez, A. Vemhet, F. Canon, P. Sami-Manchado, V. Cheynier and B. Cabane, Biophys. J., 2010, 99, 656–665 CrossRef CAS PubMed.
- A. J. Butcher, K. Gaston and P. S. Jayaraman, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 786, 3–6 CrossRef CAS.
- V. S. Rao, K. Srinivas, G. N. Sujini and G. N. S. Kumar, Int. J. Proteomics, 2014, 2014, 1–2 CrossRef PubMed.
- B. J. Mayer, J. Cell Sci., 2001, 114, 1253–1263 CAS.
- M. J. Macias, S. Wiesner and M. Sudol, FEBS Lett., 2002, 513, 30–37 CrossRef CAS PubMed.
- M. Sudol, H. I. Chen, C. Bougeret, A. Einbond and P. Bork, FEBS Lett., 1995, 369, 67–71 CrossRef CAS PubMed.
- M. Sudol and T. Hunter, Cell, 2000, 103, 1001–1004 CrossRef CAS PubMed.
- H. I. Chen, A. Einbond, S. J. Kwak, H. Linn, E. Koepf, S. Peterson, J. W. Kelly and M. Sudol, J. Biol. Chem., 1997, 272, 17070–17077 CrossRef CAS PubMed.
- H. I. Chen and M. Sudol, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7819–7823 CrossRef CAS.
- O. Ferrigno, F. Lallemand, F. Verrecchia, S. L'Hoste, J. Camonis, A. Atfi and A. Mauviel, Oncogene, 2002, 21, 4879–4884 CrossRef CAS PubMed.
- E. K. Koepf, H. M. Petrassi, M. Sudol and J. W. Kelly, Protein Sci., 1999, 8, 841–853 CrossRef CAS PubMed.
- B. G. D. la Torre, A. Jakab and D. Andreu, Int. J. Pept. Res. Ther., 2007, 13, 265–270 CrossRef.
- A. K. Tickler, A. B. Clippingdale and J. D. Wade, Protein Pept. Lett., 2004, 11, 377–384 CrossRef CAS PubMed.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
- Z. Fidan, A. Younis, P. Schmieder and R. Volkmer, J. Pept. Sci., 2011, 17, 644–649 CrossRef CAS PubMed.
- A. M. G. C. Dias, O. Iranzo and A. C. A. Roque, RSC Adv., 2015, 5, 19743–19751 RSC.
- M. Iglesias-Bexiga, F. Castillo, E. S. Cobos, T. Oka, M. Sudol and I. Luque, PLoS One, 2015, 10, e0113828 Search PubMed.
- P. A. Dalby, R. H. Hoess and W. F. DeGrado, Protein Sci., 2000, 9, 2366–2376 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- X. Daura, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 1998, 19, 535–547 CrossRef CAS.
- W. DeLano, CCP4 Newsl. Protein Crystallogr., 2002 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- C. Gracia, A. Isidro-Llobet, L. J. Cruz, G. a. Acosta, M. Álvarez, C. Cuevas, E. Giralt and F. Albericio, J. Org. Chem., 2006, 71, 7196–7204 CrossRef CAS PubMed.
- T. E. Creighton, Protein Structure: a Practical approach, IRL Press at Oxford University, Oxford, 2nd edn, 1997 Search PubMed.
- J. M. Haigh, A. Hussain, M. L. Mimmack and C. R. Lowe, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1440–1452 CrossRef CAS PubMed.
- D. A. Wellings and E. Atherton, in Methods in Enzymology, ed. E. Fields, G. B. Academic Press, San Diego, 1997, p. 54 Search PubMed.
- S. Goutelle, M. Maurin, F. Rougier, X. Barbaut, L. Bourguignon, M. Ducher and P. Maire, Fundam. Clin. Pharmacol., 2008, 22, 633–648 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10900d |
| This journal is © The Royal Society of Chemistry 2016 |