
The peculiar roles of chloride electrolytes in BDD anode cells
Chunyong Zhang*ab,
Xiaoming Dua,
Zhefeng Zhanga and
Degang Fub
aDepartment of Chemistry, College of Science, Nanjing Agricultural University, Nanjing 210095, China. E-mail: zhangchy@njau.edu.cn; Fax: +86 25 84395207; Tel: +86 25 84395207
bState Key Laboratory of Bioelectronics, Southeast University, Nanjing 210096, China
First published on 6th July 2016
Abstract
Taking phenol as a pollutant model, the role of chloride electrolytes during the anodic oxidations was investigated in a wide range of concentrations (0.1–1000 mM). Experiments were performed with an electrochemical cell containing a boron-doped diamond (BDD) anode and Pt cathode. The influence of the initial chloride concentration on the degradation performance was assessed by comparing both phenol and TOC removal efficiencies at various time intervals. The results revealed that the chloride ions played interesting and important roles during the electrolytic oxidations, and the reaction kinetics and by products might vary from case to case. Thus, we emphasize the importance of operating level selections in BDD technology, especially when high concentrations of chloride electrolytes are present in the bulk solutions.
1. Introduction
Environmental application of boron-doped diamond (BDD) has created great interest over the past decade. Specifically, the application of BDD for wastewater treatment seems a promising approach to attain real applications, due to its strong capability to mineralize biorefractory organics by electro-generated hydroxyl radicals (˙OH).1 It has been well proved that the efficiency of BDD technology depends dramatically on the adopted supporting electrolytes.2,3 So far, many electrolyte salts have been tested such as Na2SO4, NaCl, NaNO3, Na2CO3, Na3PO4 and NaClO4. Among them, NaCl is regarded as the most fascinating one due to the involvement of active chlorine. Note that in BDD anode cells, the coexistence of active chlorine species (ACS, i.e. dissolved Cl2, HClO and ClO−) and reactive oxygen species (ROS, i.e. ˙OH, ˙O2−, ˙HO2−, H2O2 and O3) will result in a complex system which does not easily allow to predict the reaction kinetics involved.4 In this scenario, relevant results concerning the influence of chloride electrolytes on the performance of BDD technology have been published. However, the results recorded in existing literature are diverse or even contradictory.5–10 For example, Aquino et al. investigated the oxidation of Reactive Red 141 dye using BDD technology.8 The effect of NaCl concentration ([NaCl], 0–2.34 g dm−3) on the system's performance was examined, indicating that higher decolorization was favored at the higher [NaCl] (>1.5 g dm−3). Conversely, efficient TOC removal was attained at lower [NaCl] (<0.7 g dm−3). In another work, they found that the addition of NaCl (1.5 g dm−3) resulted in total COD removal of a real textile effluent.9 Besides, a similar study by Zhao et al. demonstrated that NaCl played negative roles in the degradation and mineralization of diclofenac.10 Regarding the complex role of NaCl, some researchers have pointed out that the effect of active chlorine favours the formation of chlorinated intermediates, which are likely to exert an increased or a decreased persistence to further oxidation than their non-chlorinated analogues.11,12 In other words, the stability of the chlorinated byproducts under the attack of ˙OH may be of vital importance in determining the mineralization efficiency of BDD technology. On the other hand, the complex effects mentioned above are also linked to the differing ranges of [NaCl] employed, since the effect of chloride ions toward the degradation efficiency depends strongly their initial concentrations.7 Unfortunately, up to now a correlative work is still lacking in elucidating the role of chlorides in a wide range of initial chloride concentrations (e.g., 0.1–1000 mM).As a result, the exact role of chloride electrolytes in BDD anode cells is still not very clear. Thus, we report herein on a wide-range investigation of how initial chloride concentrations affect the degradation and mineralization performance of BDD technology. Phenol, a highly toxic compound widely present in industrial processes, has been chosen as a pollutant model. Note that phenol is also an important intermediate in the degradation pathway of aromatic compounds (of higher molecular weight), thus it is regarded as an ideal pollutant model for advanced wastewater treatment.13
2. Experimental
2.1. Reagents and materials
Reagent-grade phenol (C6H6O), NaCl and KH2PO4 were purchased from Wako (Japan). Deionized water was used for all stock solutions. Nb/BDD anode and Nb/Pt cathode were both from Condias (Germany).2.2. Analytical apparatus and calculations
The substrate conversion during the anodic oxidation of phenol was monitored by HPLC on an Aligent 1200 series, using a reversed-phase Kromasil C18 column. The UV detection wavelength was set as 256 nm. The mobile phase consisted of CH3OH/H2O (68/32, v/v) with a flow rate of 0.6 ml min−1. Under these conditions phenol eluted at tR of 7.3 min. The TOC values of the solutions were analyzed with a Shimadzu TOC-L analyzer to assess the efficiency of mineralization. The UV-vis and LC/MS measurements were performed using Shimadzu UV-1800 spectrophotometer and Waters Acquity UPLC/SQD analyzer, respectively.The electrochemical measurements were performed in a conventional three-electrode electrochemical cell. BDD, Pt and saturated calomel electrode (SCE) were used as working electrode, counter electrode and reference electrode, respectively. During the tests, the exposed surface area of BDD was 4.0 mm2. All measurements were made at 20 ± 1 °C and under nitrogen atmosphere.
The initial limiting current density (jlim,0) was calculated using the following equations:14
| jlim,0 = 4FkmCOD0 | (1) |
| γ = jappl/jlim,0 | (2) |
Besides, the maximum chlorine generation under mass-transfer control conditions at a planar anode (such as BDD) could be described by eqn (3).15
| jL = Fkm[NaCl] | (3) |
2.3. Degradation experiments
The degradation experiments were carried out in an undivided reactor in batch mode. BDD and Pt plates were used as anode and cathode, respectively. The effective surface areas of both electrodes were 77.44 cm2 and the electrode gap was 10 mm. For all entries, the solution volume was 500 ml and the initial concentration of phenol was 50 mg dm−3. The reaction medium was pumped through the anode system and then returned to reservoir for recycling. Samples were collected at preset time intervals to track the concentration of phenol and TOC. Note that the selections of some operating levels, such as flow rate of 400 ml min−1 and initial phenol concentration of 50 mg dm−3, were made according to those obtained in previous works.13,16 In addition, in the present work the role of chlorides was examined at two different jappl values (2.58 and 6.46 mA cm−2) to evaluate their impacts toward phenol degradation.3. Results and discussions
The results obtained for the degradation and mineralization of phenol at two different jappl values are given in Fig. 1 and 2, respectively. Some phenomena may thus be observed: | ||
| Fig. 1 Effects of initial chloride concentration on the degradation of phenol by BDD technology. ([Phenol]0: 50 mg dm−3; flow rate: 400 ml min−1; jappl: 2.58 mA cm−2 (the 1st column) and 6.46 mA cm−2 (the 2nd column)). | ||
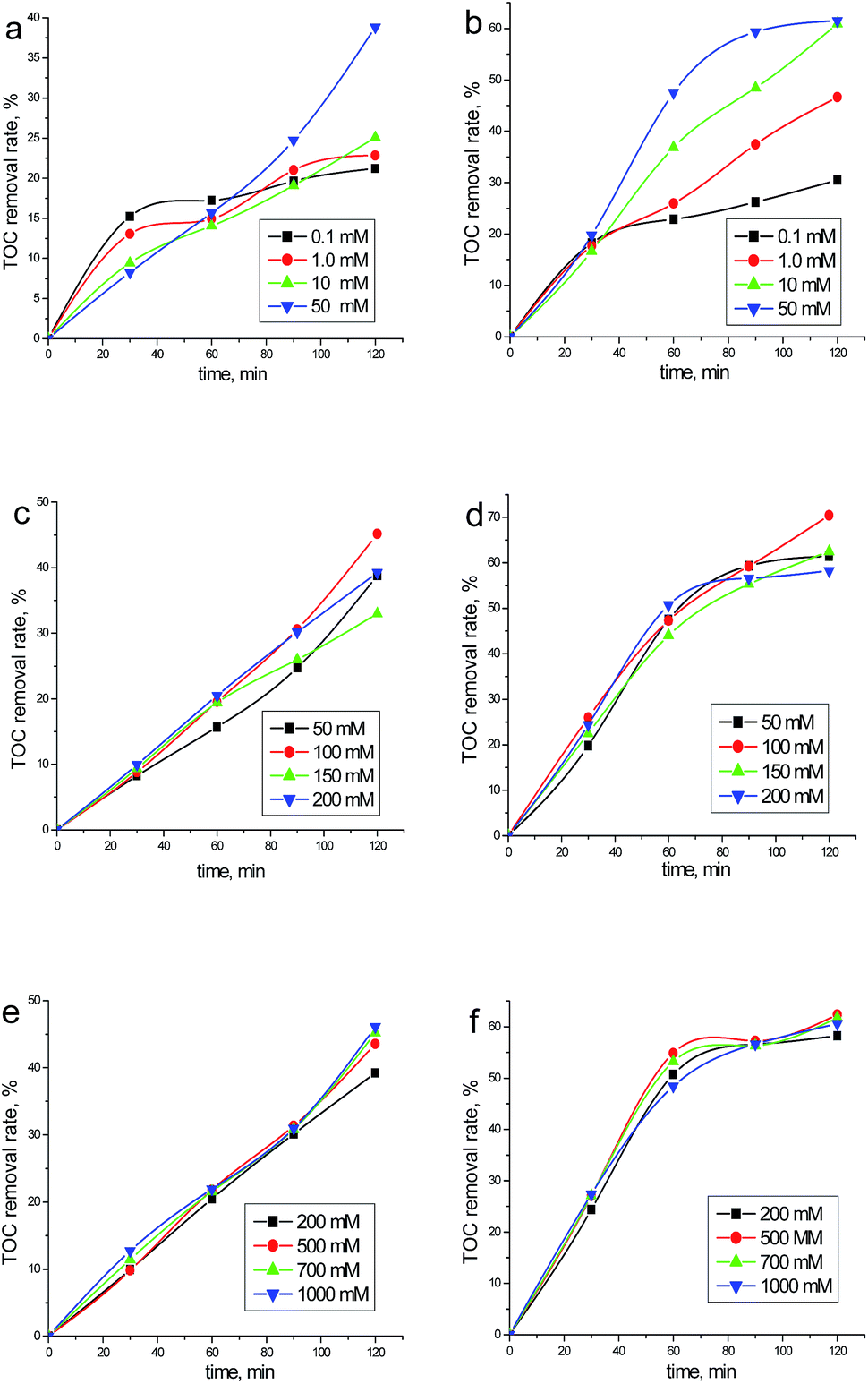 | ||
| Fig. 2 Effects of initial chloride concentration on the TOC removal rate of phenol by BDD technology. ([Phenol]0: 50 mg dm−3; flow rate: 400 ml min−1; jappl: 2.58 mA cm−2 (the 1st column) and 6.46 mA cm−2 (the 2nd column)). | ||
(1) The effect of NaCl is strongly dependent on the employed operating variables, such as jappl, [NaCl] and electrolysis time. Interestingly, [NaCl] demonstrates negative effects for a few cases (e.g., Fig. 2a).
(2) The phenol removal rate is always higher than the TOC removal rate, indicating that part of phenol has been oxidized to soluble intermediates rather than totally oxidized to CO2 and H2O. This is not surprising since the removal of phenol can be attained by either ACS or ROS, while for TOC removal, more rigorous conditions are needed to promote the production of ROS.17
(3) Regarding phenol removal, the removal efficiency increases continuously with rising [NaCl] (below 50 mM). Beyond this value, the increasing [NaCl] has no obvious promotion to the removal efficiency.
(4) In the case of phenol removal, most entries may be modeled according to pseudo-first order kinetics, indicating that active chlorine is the main oxidizing agent in the system. This is reasonable since for case of chloride mediation, the degradation process may be considered as a bi-molecular reaction between phenol and oxidant (Cl2, of a constant yield). While for a few cases (i.e. 0.1 mM NaCl), the peculiar kinetics recorded may be explained by the competitive reactions of ACS and ROS.
(5) Regarding TOC removal, it is difficult to find the relationship between removal efficiency and [NaCl] (below 50 mM). Beyond this value, the increasing [NaCl] has no obvious promotion to the mineralization efficiency.
(6) In the case of TOC removal, different reaction kinetics have been recorded. Reaction rates in Fig. 2c and e (<120 min) and Fig. 2d and f (<60 min) are very close to each other, which can be considered as zeroth order in TOC concentration. Interestingly, a saturation plateau is recorded in Fig. 2d and f (>60 min), confirming the formation of refractory intermediates. While for other cases (<50 mM [NaCl]), the mineralization kinetics involved are rather complex.
To further interpret the results, please refer to the following points:
(1) The high oxygen overpotential of BDD anode allows efficient generation of ROS (mainly ˙OH) through water electrolysis (eqn (4)). Conversely, the generation of ACS is inefficient on BDD anodes.18 Note that ACS is less reactive than ˙OH (Eθ(HOCl/Cl−) = 1.48 V, Eθ(˙OH/OH−) = 2.80 V). In BDD anode cells, a fraction of ˙OH may even be scavenged by Cl− and converted to less reactive ACS (eqn (5)).1 However, formation of ACS is highly [NaCl]-dependent. At lower [NaCl], chloride ions play minor roles because the forward and backward rates of eqn (5) are similar. In contrast, at elevated [NaCl], the forward reaction becomes dominant and the scavenging effect of chloride ions will be significant;
| BDD + H2O = BDD(˙OH) + H+ + e− | (4) |
| BDD(˙OH) + Cl− = BDD + 1/2Cl2 + OH− | (5) |
(2) The mineralization efficiency of BDD technology strongly depends on the adopted γ values, whatever the combination of the reaction conditions which generate the special γ values.19 Specifically, a mixed first and zero order kinetics will be obtained at lower γ values (i.e. <4).20 Meanwhile, the generation of active chlorine is effective only under operating conditions of higher [NaCl] (e.g., >1 mM) and higher γ values (e.g., >1).
(3) According to eqn (3), the jL values in our case may be higher or lower than the jappl values (2.58 and 6.46 mA cm−2) employed. In case these two values are equal, the [NaCl] required are 26 mM and 67 mM, respectively. Hence, under elevated chloride concentration sufficient ACS would have been generated to oxidize the substrate and this might not have been the case at lower ones. As a result, similar phenol and TOC removal rates will be obtained at elevated chloride concentrations, which are in line with several studies earlier reported.15,21
(4) An interesting observation from Fig. 2a is that at initial stage of electrolysis (i.e. 30 min), the mineralization rate decreases with rising chloride concentration. While for the case of higher jappl, the corresponding removal rates are almost the same (Fig. 2b). These phenomena may be associated with the formation and accumulation of the chlorinated intermediates, as one may conclude from the following mechanistic study.
(5) For most cases, the oxidation reactions in BDD anode cells mainly occur homogeneously in the bulk solution (through active chlorine pathways). Thus, generation of active chlorine is of particular concern. With this in mind, linear sweep voltammetry (LSV) has been adopted to measure the efficiency toward the evolution of active chlorine on BDD anodes.18 In such tests, the chlorine evolution capability is evaluated by the current difference (IT − IB) magnitude, where IT and IB refer to the current densities recorded in the presence and in the absence of chlorides, respectively. As can be seen in Fig. 3, the (IT − IB) magnitude recorded well illustrates the positive effect of [NaCl] towards chlorine evolution.
 | ||
| Fig. 3 Comparison of voltammetric currents associated with the evolution of active chlorine on BDD anode in the presence of NaCl of different initial concentrations (base electrolyte: 0.2 M KH2PO4, scan rate: 50 mV s−1). | ||
(6) In the case of lower chloride concentration, both ROS and ACS play major roles during the electrolytic oxidations. While for case of higher ones, the role of ROS becomes increasingly buffered by the chloride electro-activity, and the formation of ˙Cl species should then be taken into account, alongside with other ACS generated on BDD surface.21 As a consequence, some inorganic byproducts such as chlorate (ClO3−) and perchlorate (ClO4−) may be generated during the electrolysis, which is detrimental to the degradation of substrate.22,23
Based on the above analyses, it can be concluded that generated intermediates will vary greatly with changing initial chloride content. Here, we would like to draw a special attention to the work of Chen et al. where they found a striking difference concerning the chlorinated oligomers when different [NaCl] values (i.e. 5 mM and 50 mM) were employed.24 Specifically, Chen and his co-workers had found the formation of dimers and trimers of phenol (as important intermediates) in the case of higher [NaCl]. Considering that a wider [NaCl] range has been adopted in the present study, a similar mechanistic investigation will surely offer useful information on the chloride effects.
Prior to studying the degradation intermediates, UV-vis measurements were conducted to obtain additional information about the chloride effects in the electro-oxidation processes of phenol. As shown in Fig. 4, five representative UV-vis spectra are presented with 0.1, 1.0, 10, 100 and 1000 mM NaCl as the supporting electrolytes, respectively. Specifically, in the case of low [NaCl], the adsorption peaks of phenol (212 and 269 nm) both decrease with the proceeding of electrolysis, confirming that hydroxyl radicals are the main oxidants in the system (Fig. 4a and b). When [NaCl] reaches 10 mM, two new peaks (242 and 315 nm) arise at initial stage of degradation (i.e. 30 min), while the peak at 269 nm vanishes. Thereafter, these peaks decrease quickly with electrolysis time (Fig. 4c). This phenomenon may be attributed to the generation/consumption process of the aromatic intermediates, as well as to the competitive reactions of ACS and ROS. For cases of 100 and 1000 mM NaCl, the broad adsorption band generated around 280 nm (in Fig. 4d and e) is indicative of the formation of chlorinated organic intermediates, chlorate and perchlorate.25 These results can be linked to the enhanced contribution of ACS, as well as to the intensified ClO3− and ClO4− formation reactions on BDD surface.
 | ||
| Fig. 4 UV-vis spectra changes of phenol in NaCl media (of different initial concentrations) during the electrocatalytic oxidation on BDD anode ([Phenol]0: 50 mg dm−3; flow rate: 400 ml min−1; jappl: 6.46 mA cm−2). | ||
As a result, the different patterns recorded in these graphs evidence that chloride concentration does impact intermediate speciation. It is also reasonable to assume that more chlorinated intermediates will be formed at elevated chloride concentrations.
To confirm the hypothesis, during the anodic oxidations by applying a jappl of 6.46 mA cm−2, five samples were all collected at 120 min and analyzed by the LC/MS technique. Briefly, the analyses permitted the identification of eighteen main intermediates, compounds with m/z value of 108, 110, 110, 126, 128, 128, 142, 144, 163, 186, 197, 220, 234, 239, 285, 289, 321 and 324.
From the results obtained hereinbefore, the proposed degradation pathways for phenol oxidation in BDD/Cl− system are shown schematically in Fig. 5. It is assumed that Cl2 and ˙OH are the main oxidizing agents in the reaction system.
 | ||
| Fig. 5 Proposed degradation sequence of phenol during BDD anodic oxidations in the presence of NaCl. | ||
According to the scheme, under the attack of the oxidants, the phenol degradation is found to follow oxidation pathway leading to hydroxylated and chlorinated intermediates through competitive routes. Specifically, one important step consists of an electrophilic substitution leading to chlorinated byproducts, i.e. chlorine attack at the ortho- and para-positions of phenol to form aromatic chlorohydrocarbon. The oxidation process may continue with a cleavage of aromatic rings, resulting in the production of organic acids.26,27 As expected, the distribution of the main reaction intermediates is closely linked the adopted initial chloride concentrations (see Table 1).
| Intermediates | 0.1 mM | 1.0 mM | 10 mM | 100 mM | 1000 mM |
|---|---|---|---|---|---|
| 1 (m/z 110) | √ | √ | |||
| 2 (m/z 110) | √ | √ | |||
| 3 (m/z 126) | √ | √ | √ | ||
| 4 (m/z 142) | √ | √ | |||
| 5 (m/z 108) | √ | √ | |||
| 6 (m/z 128) | √ | √ | √ | √ | √ |
| 7 (m/z 128) | √ | √ | √ | √ | √ |
| 8 (m/z 163) | √ | √ | √ | √ | |
| 9 (m/z 197) | √ | √ | √ | √ | |
| 10 (m/z 144) | √ | √ | √ | √ | |
| 11 (m/z 186) | √ | √ | √ | ||
| 12 (m/z 220) | √ | √ | √ | ||
| 13 (m/z 239) | √ | √ | √ | ||
| 14 (m/z 285) | √ | √ | |||
| 15 (m/z 234) | √ | ||||
| 16 (m/z 289) | √ | √ | √ | ||
| 17 (m/z 324) | √ | ||||
| 18 (m/z 321) | √ |
As a whole, it can be concluded that the following factors are mainly responsible for the peculiar roles of chloride electrolytes recorded in BDD anode cells: (1) the electrolysis time; (2) the initial chloride concentration (or jL value) employed; (3) the jappl value (or γ value) employed; (4) the respective reactivity of ROS and ACS with substrate; (5) the respective reactivity of ROS and ACS with the degradation intermediates (chlorinated or non-chlorinated ones) and (6) the competitive formation reactions of chlorate and perchlorate. Considering the possible environmental risks of the chlorinated intermediates, experiments with a special emphasis on evaluating their toxicity should be carried out in future studies.
4. Conclusions
In this investigation we have attempted to elucidate the role of chlorides in BDD anode cells. Our findings indicate that, the effects of chloride electrolytes toward the degradation efficiency depend strongly on the adopted reaction conditions. Moreover, different chlorinated phenolic oligomers will be formed with varying initial chloride concentrations. This is the first wide-range study that elucidates the role of chloride ions in anodic oxidation processes, which should be taken into consideration when such technology is applied to the treatment of chloride-containing wastewaters.Acknowledgements
This study has received funding from the Fundamental Research Funds for the Central Universities (KYZ201648), and the Open Research Fund of the State Key Laboratory of Bioelectronics, Southeast University under the grant agreement No. 2016B08.References
- C. A. Martinez-Huitle, M. A. Rodrigo, I. Sires and O. Scialdone, Chem. Rev., 2015, 115, 13362–13407 CrossRef CAS PubMed.
- J. M. Aquino, M. A. Rodrigo, R. C. Rocha-Filho, C. Sáez and P. Cañizares, Chem. Eng. J., 2012, 184, 221–227 CrossRef CAS.
- C. Zhang, Z. He, J. Wu and D. Fu, J. Electrochem. Soc., 2015, 162, E85–E89 CrossRef CAS.
- O. Scialdone, S. Randazzo, A. Galia and G. Silvestri, Water Res., 2009, 43, 2260–2272 CrossRef CAS PubMed.
- B. N. Grgur and D. Mijin, Appl. Catal., B, 2014, 147, 429–438 CrossRef CAS.
- C. Zhang, L. Liu, W. Li, J. Wu, F. Rong and D. Fu, J. Electroanal. Chem., 2014, 726, 77–83 CrossRef CAS.
- G. R. P. Malpass, D. W. Miwa, D. A. Mortari, S. A. S. Machado and A. J. Motheo, Water Res., 2007, 41, 2969–2977 CrossRef CAS PubMed.
- J. M. Aquino, R. C. Rocha-Filho, M. A. Rodrigo, C. Saez and P. Canizares, Water, Air, Soil Pollut., 2013, 224, 1397–1406 CrossRef.
- J. M. Aquino, G. F. Pereira, R. C. Rocha-Filho, N. Bocchi and S. R. Biaggio, J. Hazard. Mater., 2011, 192, 1275–1282 CrossRef CAS PubMed.
- X. Zhao, Y. Hou, H. Liu, Z. Qiang and J. Qu, Electrochim. Acta, 2009, 54, 4172–4179 CrossRef CAS.
- M. Wu, G. Zhao, M. Li, L. Liu and D. Li, J. Hazard. Mater., 2009, 163, 26–31 CrossRef CAS PubMed.
- A. M. S. Solano, C. K. C. Araujo, J. V. Melo, J. M. Peralta-Hernandez, D. R. Silva and C. A. Martinez-Huitle, Appl. Catal., B, 2013, 130–131, 112–120 CrossRef.
- J. Wei, X. Zhu and J. Ni, Electrochim. Acta, 2009, 56, 5310–5315 CrossRef.
- M. Panizza and G. Cerisola, Chem. Rev., 2009, 109, 6541–6569 CrossRef CAS PubMed.
- I. Pikaar, R. A. Rozendal, Z. Yuan, J. Keller and K. Rabaey, Water Res., 2011, 45, 5381–5388 CrossRef CAS PubMed.
- C. Zhang, J. Wang, T. Murakami, A. Fujishima, D. Fu and Z. Gu, J. Electroanal. Chem., 2010, 638, 91–99 CrossRef CAS.
- L. Liu, B. Li, Z. He, C. Zhang and D. Fu, Sep. Purif. Technol., 2015, 146, 15–23 CrossRef CAS.
- J. Joonseon, K. Choonsoo and Y. Jeyong, Water Res., 2009, 43, 895–901 CrossRef PubMed.
- M. Mascia, A. Vacca, A. M. Polcaro, S. Palmas, J. R. Ruiz and A. D. Pozzo, J. Hazard. Mater., 2010, 174, 314–332 CrossRef CAS PubMed.
- S. Li, D. Bejan, M. S. McDowell and N. J. Bunce, J. Appl. Electrochem., 2008, 38, 151–159 CrossRef CAS.
- J. H. B. Rocha, M. M. S. Gomes, E. V. Santos, E. C. M. Moura, D. R. Silva, M. A. Quiroz and C. A. Matinez-Huitle, Electrochim. Acta, 2014, 140, 419–426 CrossRef CAS.
- M. E. H. Bergmann and J. Rollin, Catal. Today, 2007, 124, 198–203 CrossRef CAS.
- O. Azizi, D. Hubler, G. Schrader and B. P. Chaplin, Environ. Sci. Technol., 2011, 45, 10582–10590 CrossRef CAS PubMed.
- L. Chen, P. Campo and M. J. Kupferle, J. Hazard. Mater., 2015, 283, 574–581 CrossRef CAS PubMed.
- M. Santhanam, S. Annamalai, S. Sudanthiramoorthy and R. Gopalakrishnan, RSC Adv., 2015, 5, 75528–75532 RSC.
- Y. Zhang, N. Yang, M. Murugananthan and S. Yoshihara, J. Hazard. Mater., 2015, 244–245, 295–302 Search PubMed.
- Y. Ji, C. Dong, D. Kong and J. Lu, J. Hazard. Mater., 2015, 285, 491–500 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |