
Unimolecular micelles from graft copolymer with binary side chains
Abstract
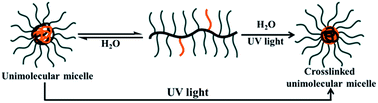
A novel amphiphilic binary graft copolymer poly(glycidyl methacrylate)-graft-[poly(2-cinnamoyl-oxyethyl methacrylate)-random-methoxy polyethylene glycol] (PGMA-g-(PCEMA-r-MPEG)) was successfully synthesized by a combination of atom transfer radical polymerization (ATRP) and click reaction, in which alkyne-end-functionalized poly(2-cinnamoyloxyethyl methacrylate) (PCEMA–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) and poly(ethylene glycol) methyl ether (MPEG–CCH) were grafted onto a poly(3-azide-2-hydroxy-propyl methacrylate) (P(GMA-N3) backbone. This polymer was used to prepare stable unimolecular micelles (UMMs), which could be produced using either high or low polymer concentrations. Since water is a good solvent only for MPEG but a poor solvent for both PGMA and PCEMA, the hydrophobic PGMA and PCEMA segments aggregated together to form a dense core that was surrounded by a corona based on the soluble MPEG segments. PCEMA was photo-crosslinkable, and thus the UMMs could be crosslinked by shining UV light on the system to yield permanent UMMs. The morphologies of the UMMs were characterized by TEM, AFM, and DLS. Both the TEM and AFM observations indicated that the crosslinked UMMs had a diameter of ∼13 nm, while the DLS measurements indicated they had a diameter of ∼34 nm. The unimolecular state of the micelles was confirmed by SEC, as well as a comparison of the theoretical mass per graft copolymer molecule with that of an individual micelle. Moreover, the morphology of the UMMs was unperturbed by the crosslinking reaction although they became more compact and had a slightly smaller diameter.
CH) and poly(ethylene glycol) methyl ether (MPEG–CCH) were grafted onto a poly(3-azide-2-hydroxy-propyl methacrylate) (P(GMA-N3) backbone. This polymer was used to prepare stable unimolecular micelles (UMMs), which could be produced using either high or low polymer concentrations. Since water is a good solvent only for MPEG but a poor solvent for both PGMA and PCEMA, the hydrophobic PGMA and PCEMA segments aggregated together to form a dense core that was surrounded by a corona based on the soluble MPEG segments. PCEMA was photo-crosslinkable, and thus the UMMs could be crosslinked by shining UV light on the system to yield permanent UMMs. The morphologies of the UMMs were characterized by TEM, AFM, and DLS. Both the TEM and AFM observations indicated that the crosslinked UMMs had a diameter of ∼13 nm, while the DLS measurements indicated they had a diameter of ∼34 nm. The unimolecular state of the micelles was confirmed by SEC, as well as a comparison of the theoretical mass per graft copolymer molecule with that of an individual micelle. Moreover, the morphology of the UMMs was unperturbed by the crosslinking reaction although they became more compact and had a slightly smaller diameter.
 Please wait while we load your content...
Please wait while we load your content...

