DOI:
10.1039/C6RA10491F
(Paper)
RSC Adv., 2016,
6, 74215-74224
Hydrogen desorption behaviour and microstructure evolution of a γ-AlH3/MgCl2 nano-composite during dehydriding†
Received
22nd April 2016
, Accepted 27th July 2016
First published on 1st August 2016
Abstract
A γ-AlH3/MgCl2 nano-composite, without or with Zn and Zr doping, was synthesized by solid state reaction milling using MgH2 and AlCl3 as reagents. The hydrogen desorption behaviour of the nano-composite was investigated by temperature programmed de-hydriding (TPD) and differential scanning calorimetry (DSC) tests, and the microstructure evolution due to de-hydriding was characterized by XRD, SEM, and TEM respectively. For de-hydriding by heating from 40 to 320 °C, a three-stage featured TPD curve was observed, with the maximum hydrogen desorption capacity excluding the MgCl2 in the composite achieving about 9.71 wt% when the temperature was raised to 240 °C and then remaining unchanged for the subsequent heating process to 320 °C. DSC and XRD tests revealed that three individual events, i.e., the transformation of γ-AlH3 to α-AlH3 phase, the direct decomposition of γ-AlH3 phase, and the subsequent decomposition of the α-AlH3 phase, take place during the de-hydriding process of the γ-AlH3/MgCl2 nano-composite. By doping the nano-composite with elements Zn and Zr, an improvement in the de-hydriding kinetics was observed. According to the calculation based on the fitting of de-hydriding kinetics data, it was found that the addition of Zn and Zr can reduce the activation energy for the de-hydriding reaction. Also, both the de-hydriding mechanism and the role that Zn and Zr play in the de-hydriding process were interpreted based on TEM observations.
1. Introduction
As a new and green energy source, hydrogen is regarded as a perfect carrier to replace fossil fuels for such applications as energy storage and hydrogen-fueled cells due to its unique features of non-toxicity, high energy density and promising performance in fuel cells.1–5 In recent years, on-board hydrogen storage has posed considerable technical challenges that could be detrimental to the application of fuel cells.6,7 Therefore, a lightweight, effective and high capacity hydrogen storage material should be developed for hydrogen storage.8–12 Among the various light metal hydrides, AH3 (alane) with a high gravimetric hydrogen capacity of 10.1 wt%, a relatively low desorption temperature (100–200 °C) and a minor dehydriding enthalpy, is acknowledged as a fascinating material, and has attracted more and more attention for its potential as a promising hydrogen storage candidate.13–18
Non-solvated AlH3 with seven variations in its crystal structures, i.e., α, α′, β, γ, δ, ε and ζ phase, was firstly synthesized by the direct reaction of LiAlH4 and AlCl3 in diethyl ether solvent, as shown in reaction (1).19 Due to the unique structure and different Al–H arrangement, each phase has different thermodynamic and dehydriding properties.20 Based on the thermodynamic calculation, it is deduced that the α and other phases of AlH3 can decompose spontaneously at room temperature.21–24 But, it was demonstrated by several groups that the above dehydriding reaction can not occur due apparently to the stable oxide layer which can protect the AlH3 from decomposing at ambient temperature.25–29 Wang et al. found that the rate of hydrogen release through the oxide barrier was limited with increasing thickness of Al2O3 layer.26 Kato et al. has clearly demonstrated that the channels formed in the oxide is associated with the break-up of the oxide layer due to the greater thermal expansion of AlH3 compared to that of Al2O3 at elevated temperature for dehydriding.28 Therefore, the thickness of surface oxide is regarded as an effective factor for AlH3 stabilization at room temperature. However, it was verified by Graetz that the decomposition of fresh AlH3 polymorphs (without any oxide layer on the surface) is still not feasible at room temperature mainly due to the effect of dehydriding kinetics, and the thermal decomposition kinetics is significantly controlled by nucleation and growth of Al.29
| | |
3LiAlH4 + AlCl3 + Et2O → 4AlH3 + 3LiCl↓ + Et2O↑
| (1) |
Among the seven AlH3 polymorphs, the unit cell, lattice parameters and bridge bonds of the α phase were determined by Turley et al. in 1969, in which only one type of corner connected AlH6 octahedra exists.30 Compared with the most valued α-AlH3, the structure of γ-AlH3 with an orthorhombic unit cell was not determined or thoroughly investigated by Yartys until 2007.31 The unique bifurcated double-bridge bonds (Al–2H–Al) in the AlH6 octahedra building blocks result in large cavities between AlH6 octahedra.32 In view of the above structural analysis, γ-AlH3 is less stable than the α phase. Although Brower found that γ-AlH3 can transform into α phase by the desolvation of AlH3·Et2O and subsequently heating treatment (see reaction (2)), more details about the thermal property of γ phase, such as the reaction mechanism and transition enthalpy of the conversion, was not discussed by Graetz until 2006,24 from which the γ to α phase transition enthalpy at 298 K were calculated to be 2.8 kJ mol−1, based on DSC analysis. It was later calculated by Orimo et al. that the Gibbs energy for formation of α phase at 300 K turns out to be about −31 kJ mol−1 H2.33,34 Therefore, it can be concluded that α-AlH3 is thermodynamically recognized to be more stable than other types of AlH3 phases,24,35 and the phase transition to α-AlH3 is possible if the atoms can obtain enough energy to overcome the energy barrier for structure reform.
| | |
AlH3·nEt2O → γ-AlH3 + nEt2O↑ → α-AlH3
| (2) |
Sandrock demonstrated that during the process of dehydrogenation, the decomposition of α-AlH3 with a large particle (50–100 μm) or with a thicker oxide layer needs a higher dehydriding temperature (150–200 °C).13 Even if the γ-AlH3 was prepared and used immediately, it was also verified by Liu that the initial dehydriding temperature is still to be about 130 °C,7 which probably hinders the application of AlH3 as a hydrogen storage material. A promising approach proposed by Gutowska shows that the nano-sized metal hydride could increase the dehydriding kinetics without doping of catalyst.36 Thus, numerous searching groups focused on improving the desorption kinetics by using a ball milling method.37–39 Orimo et al. found that milling the as-prepared AlH3 could reduce the dehydriding temperature and accelerate the desorption rates.34 But, their efforts were mainly focused on the thermodynamics of milled AlH3, not the kinetic and mechanism of the dehydriding reaction. It was also reported by Sandrock that the dopant of alkali metal hydrides could increase low-temperature H2 desorption kinetics if the grain size of AlH3 is finally controlled in an appropriate range.40 For example, with the addition of LiH, NaH, KH into the as-milled AlH3, the dehydriding temperature could be reduced by 25–50 °C. Similarly, Graetz reported that the nano-sized AlH3 synthesized by wet chemical method also exhibits an desirable decomposition temperature less than 100 °C,20 and has a high H2 yield which can approach the theoretical hydrogen content of AlH3 (10 wt%). It is also noted that nano-sized AlH3 can be obtained without needing furthermore ball milling or mixing as-prepared AlH3 with small levels of the alkali metal hydrides.29 Nevertheless, the above methods are not impractical for AlH3 mass production. Namely, an additional approach such as ball milling should be needed to get the final nano-sized product from the as-synthesized AlH3. Furthermore, the desolvating process for removing large quantities of organic solvents from the solvates is uneconomical and hazardous.
As an alternative route, the mechanochemical synthesis is considered to be a green and economical method to obtain metal hydrides.41–43 In the research of mechanical decomposition of LiAlH4 with VCl3, VBr3 and AlCl3, it was found by Fernandez that a mechanochemical reaction between LiAlH4 and AlCl3 occurred and AlH3 was obtained during milling.43 Thus, this mechano-chemical method was investigated and used in a desirable way to synthesize nano-sized AlH3 by several research groups.44–48 For example, Paskevicius et al. discovered that several polymorphs of AlH3 (α-, α′-, β-, and γ-AlH3) can be obtained with a solid state reaction between LiAlH4 and AlCl3 at low (77 K) or ambient temperature, and the final grain size of the AlH3 can be as fine as 16 nm.45 Nevertheless, as a promising hydrogen storage medium, the dehydriding property of as-milled AlH3–chloride composite has not been investigated systematically yet, and therefore more effort needs to focus on the mechanism, dehydriding kinetics of AlH3-based nano-composite. In our previous work, a novel γ-AlH3/MgCl2 nano-composite was successfully prepared by a solid state reaction between nanocrystallite MgH2 and AlCl3.49 In the present study, the de-hydriding process, the microstructural evolution and dehydriding kinetics of this γ-AlH3/MgCl2 nano-composite is thoroughly investigated.
2. Experimental
The γ-AlH3/MgCl2 nano-composite was synthesized by solid state reaction milling with nanocrystallite MgH2 (self made, 99%) and AlCl3 (Sigma Aldrich, 99%) as starting materials without further purification and the amount of the loaded MgH2 and AlCl3 was stoichiometric with a mole ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 for reaction (3).49 The nanocrystallite MgH2 was prepared by milling pure Mg or Mg-5.5%Zn-0.6%Zr (mass%) alloy powders in hydrogen to full hydrogenation. The reactive ball milling of MgH2 and AlCl3 was performed by using a planetary-type QM-SP4 device fitted with four 500 cm3 canisters. During ball milling, the hydrogen pressure in the canister was kept above 1 MPa. The ball-to-powder mass ratio was 60:1, and the mill shaft rotation was 400 rpm.
:2 for reaction (3).49 The nanocrystallite MgH2 was prepared by milling pure Mg or Mg-5.5%Zn-0.6%Zr (mass%) alloy powders in hydrogen to full hydrogenation. The reactive ball milling of MgH2 and AlCl3 was performed by using a planetary-type QM-SP4 device fitted with four 500 cm3 canisters. During ball milling, the hydrogen pressure in the canister was kept above 1 MPa. The ball-to-powder mass ratio was 60:1, and the mill shaft rotation was 400 rpm.| | |
3MgH2 + 2AlCl3 → 2AlH3 + 3MgCl2
| (3) |
The as-prepared and dehydrided γ-AlH3/MgCl2 nano-composite powders were investigated by XRD analysis. The powder X-ray diffraction measurements were performed at room temperature by using a Philips X'PERT diffractometer with monochromized Cu-Kα X-radiation. The voltage and tube current were 40 kV and 40 mA, respectively. Position-sensitive detector continuously covered the 2θ with a range of 10 to 90°. To prevent the Al phase from oxidation, the dehydrided samples were processed in glove box and covered with a thin polyethylene film and exposed to air only for a few minutes during the transferring operation.
Non-isothermal dehydriding test was carried out by using a home-made vacuum apparatus with a special reaction chamber. For each run of test, 10 g of γ-AlH3/MgCl2 nano-composite was loaded into a stainless steel holder. The initial particle size of the nano-composite can be calculated based on SEM observations, as shown in Fig. S1 (refer to ESI†). Upon milling for 25 h, the average size of most individual particles was estimated to be about 1 μm. By comparing between (a) and (b) in Fig. S1,† it is evident that the particles of the γ-AlH3/MgCl2 nano-composite synthesized without dopping of Zn and Zr tend to be more agglomerated. During the temperature programmed desorption (TPD) process, the rate of heating as well as the vacuum in the reaction chamber were controlled by a computer and monitored in situ with digital vacuum gauges. To investigate the dehydriding behavior of the γ-AlH3/MgCl2 nano-composite, the TPD measurements were performed from 40 to 320 °C with a heating rate of 1 °C min−1. The hydrogen content desorbed from the composite was calculated in terms of the chamber pressure change. The amount of hydrogen was calculated by the ideal gas equation using the obtained pressure data. Based on the stoichiometric mass of the γ-AlH3 phase in the nano-composite samples and the above results, the desorption curves of the as-prepared γ-AlH3/MgCl2 nano-composites with or without Zn and Zr dopping were obtained. It should be noted that, for the as-prepared samples dopped with Zn and Zr, the calculation was based on the net mass of γ-AlH3 by excluding the mass of the dopping Zn and Zr elements in the nano-composites.
Thermal analysis was studied by differential scanning calorimetry (DSC) by using a DSC METTLER TOLEDO instrument. In order to prevent the sample from oxidizing, the sample was sealed into an aluminum crucible in glove box and quickly transferred to the instrument in T-zero pans. During the measurement, the argon was flowing constantly at 20 mL min−1 to minimize the sample exposure to air. The DSC measurements were carried out by heating the samples from 40 to 260 °C at various rates of 3, 5, 10, 15 °C min−1, respectively.
The dehydrided sample powders were coated with a thin layer of gold (Au) by sputtering (Emitech K450X), and the variation of the powder morphology due to desorption was then observed on a scanning electron microscope (SEM, Quanta 200 FEG) operating at an acceleration voltage of 20 kV. The transmission electron microscopy (TEM) study was carried out on the JEOL JEM2011 apparatus. For TEM observation, the sample powders were loaded onto the carbon support films with 200 mesh copper grids.
3. Results and discussion
3.1 XRD characterization of powder samples
The XRD patterns of both the as-prepared and the dehydrided γ-AlH3/MgCl2 nano-composite samples are presented in Fig. 1. The dehydriding experiments were carried out at various temperatures and the dehydriding duration was 6 h. An amorphous-like background in the XRD patterns between 13° and 20° is ascribed to the a polyimide film used to seal the samples. Fig. 1(a) shows the XRD patterns of the powder sample milled for 25 h, using MgH2 and AlCl3 as starting material, with ball to powder mass ratio of 60:1. This indicated that, in the final milling stage, nano-crystallite γ-AlH3 was formed. It can be seen from Fig. 1(b) that the sample dehydrided at 90 °C for 6 h contains γ-AlH3, α-AlH3 and MgCl2. Moreover, some trace of Al exists in the sample, which indicates that a part of γ-AlH3 has decomposed directly during this dehydriding process. Since there also exists some α-AlH3 in the dehydrided γ-AlH3/MgCl2 nano-composite sample, this suggests that a fraction of γ-AlH3 phase has transformed into α phase during the above dehydriding process. This result is in good accordance with the work by Liu et al. who found that the polymorph transformation from γ-AlH3 to a-AlH3 occurred at an elevated temperature of 25–100 °C.7 The remaining MgCl2 indicates that AlH3 prefers to decompose by itself and does not react with MgCl2 at 90 °C. With the dehydriding temperature increasing to 180 °C, there is a substantial change in XRD patterns of the dehydrided sample as shown in Fig. 1(c), from which only diffraction peaks of Al and MgCl2 are observed, suggesting that the full dehydriding of both γ-AlH3 and α-AlH3 has been achieved after dehydriding at this temperature for 6 h. With the dehydriding temperature enhanced further to 240 °C, no new diffraction peaks except for those of Al and MgCl2 were observed, as shown in Fig. 1(d), but the intensity of the peaks assigned to Al increased significantly. This suggests that with the dehydriding temperature increasing to 240 °C, the Al formed due to decomposition of AlH3 still does not react with MgCl2, but grain growth of the Al phase formed during dehydriding is notable at a higher dehydriding temperature of 240 °C.
 |
| | Fig. 1 XRD profiles of γ-AlH3/MgCl2 nano-composite: (a) as-prepared and (b)–(d) subsequently dehydrided for 6 h at 90 °C, 180 °C, and 240 °C respectively. | |
3.2 The non-isothermal dehydriding of the γ-AlH3/MgCl2 system
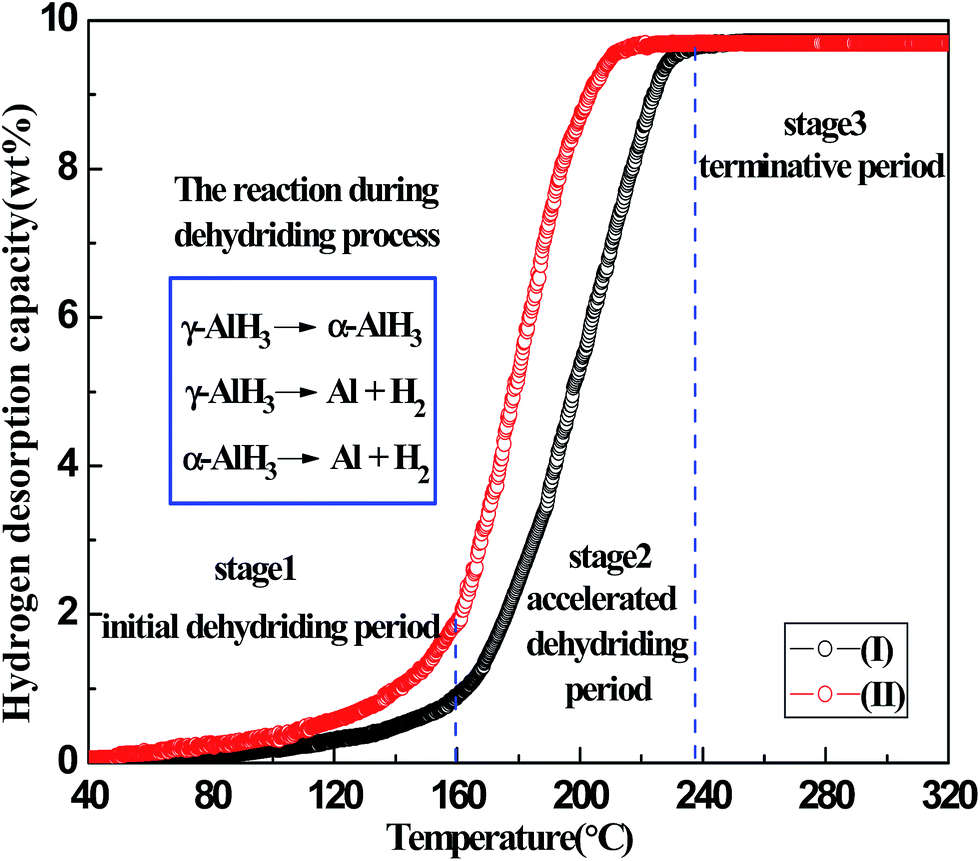
The comparative non-isothermal dehydriding curves of the γ-AlH3/MgCl2 nano-composite samples with and without Zn and Zr addition are shown in Fig. 2. The curves of non-isothermal dehydriding were obtained by the temperature-programmed-desorption (TPD) measurements from 40 to 320 °C with a heating rate of 1 °C min−1. It is noted that the hydrogen desorption weight percent can be calculated based on the sample weight and the stoichiometric weight of γ-AlH3 in the sample, respectively. As shown in Fig. S2 (refer to ESI†), the hydrogen desorption of the γ-AlH3/MgCl2 nano-composite was calculated based on the whole sample weight, in which the hydrogen desorption capacity was about 1.69 wt%, which is very close to the theoretical hydrogen capacity of the nano-composite with a stoichiometric ratio of 2AlH3 to 3MgCl2 (1.75 wt%). It is shown from Fig. 2 that the as-prepared γ-AlH3/MgCl2 nano-composite starts to release hydrogen approximately at 70 °C and has a slow desorption kinetics between 70 and 160 °C. The hydrogen released from the composite is only about 0.8 wt% in the stage when it is heated from room temperature to 160 °C. This hydrogen content is much lower than the theoretical gravimetric hydrogen capacity of AlH3, suggesting that a large proportion of AlH3 has not decomposed under a relatively lower temperature. This phenomenon is in good accordance with the above XRD analysis, in which some trace of AlH3 is also detected after isothermal dehydriding at 90 °C for even 6 h. When it is further heated at above 160 °C, with its temperature being increased gradually, the γ-AlH3/MgCl2 nano-composite presents fast dehydriding kinetics. This fast dehydriding stage starts at around 160 °C and goes to an end at about 238 °C, when a hydrogen desorption capacity of 9.71 wt%, which is very close to its theoretical hydrogen capacity of 10.1 wt%, is achieved. This should be attributed to the full decomposition of the AlH3 in the nano-composite. Thus, it is clear that the hydrogen content of the γ-AlH3/MgCl2 nano-composite comes solely from the desorption of AlH3. In accordance with the dehydriding behavior of the pure AlH3, the weight percent of the hydrogen desorbed can be calculated based on the stoichiometric AlH3 weight, and the desorption curves in Fig. 2 indicate that the γ-AlH3/MgCl2 nano-composite also exhibits a desirable and high hydrogen-storage property (the hydrogen content is 9.71 wt%). This result has a good correspondence with the report addressed by Paskevicius who found that the hydrogen desorption kinetic curves can be described by hydrogen content as a function of time in the calculated non-salt AlH3 portion in the composite.45 In overall, the TPD curves of the γ-AlH3/MgCl2 nano-composite exhibits a three-stage feature. Namely, the initial slowly dehydriding stage starts at 70 °C and ends at 160 °C, the subsequently accelerated fast dehydriding stage starting at temperature above 160 °C, and the final stage identified as the terminative period, as shown in Fig. 2. Based on the aforementioned XRD and TPD results, it can be concluded that three reactions have occurred during the heating process, as shown in eqn (4)–(6). However, Compared with the curve of (I) in Fig. 2, it is apparent that when dopped with Zn and Zr, the starting temperature for dehydriding of the nano-composite can be lowered to 50 °C. Furthermore, by comparing curves (I) and (II), it is obvious that the dehydriding rate of the γ-AlH3/MgCl2 nano-composite dopped with Zn and Zr is also faster. That is to say, AlH3 dopped with Zn and Zr presents a more desirable dehydriding kinetics, suggesting that the effect of Zn and Zr dopping on the dehydriding of AlH3 is positive. These results have good correspondence with our previous work in which Zn and Zr were found to accelerate the dehydriding reaction of AlH3.50
 |
| | Fig. 2 TPD curves of as-prepared (I) γ-AlH3/MgCl2 nano-composite and (II) γ-AlH3/MgCl2 nano-composite dopped with Zn and Zr. The hydrogen content is given as a percentage of the non-salt portion of the samples. | |
3.3 Phase transition and desorption properties
In order to get a deep insight into the dehydriding process of the γ-AlH3/MgCl2 nano-composite, further supporting evidence can be obtained from the DSC curves in Fig. 3. It has been demonstrated by previous work that there exist three DSC peaks in the dehydriding process,7,50 the exothermic peak is associated with the polymorph transformation of some γ-AlH3 to α-AlH3, and the low-temperature endothermic peak corresponds the direct decomposition of the γ-AlH3, while the high-temperature endothermic peak is related to the dehydriding of the α phase which was formed by phase transformation of γ-AlH3. As can be seen from Fig. 3, all the DSC curves obtained at various heating rates exhibit a complex but similar desorption process, and this desorption process in the temperature range of 90–190 °C also contains one exothermic peak and two endothermic peaks. It is interesting to find that a portion of γ-AlH3 decomposes directly, while the remaining γ-AlH3 first transforms to α-AlH3 and subsequently decomposes at relatively higher temperature. It is also found from Fig. 3 that the decomposition of α-AlH3 needs a relatively higher temperature than that of γ-AlH3, attributed to the more stable structure of the α phase.32 More details on the crystallite structure change of AlH3 during the dehydriding process are shown in Fig. 4. It is easy to see that the crystallite structure of γ-AlH3 with large cavities between AlH6 octahedra has a lower density than α phase. When it is heated, the γ-AlH3 can be either transformed into α-AlH3 via a phase transition reaction or decomposed directly to release H2. From Fig. S3 in ESI,† it is obvious that the desorption curves of the nano-composite dopped with Zn and Zr still have similar peaks to that of the un-doped composite. This phenomenon indicates that no new phase was formed in the Zn and Zr dopped product. Namely, no hydride of Zn or Zr was formed in the γ-AlH3/MgCl2 nano-composite. However, it is noted that, by dopping with Zn and Zr, the temperature needed for dehydriding is reduced remarkably. Thus, it can be deduced that the elements Zn and Zr have positive effects on the decomposition kinetics of AlH3. To determine the value of the apparent activation energies (Ea) for this dehydriding process, the desorption kinetics of the γ-AlH3/MgCl2 nano-composite was studied by using the Kissinger's method. Moreover, the relationship among the activation energy (Ea), the heating rate (c), and the peak temperature of dehydriding (TP) in the DSC curve can be formulated by following Kissinger's equation:| | |
ln(c/Tp2) = −(Ea/RTp) + A
| (7) |
 |
| | Fig. 3 DSC curves of γ-AlH3/MgCl2 nano-composite at various heating rates. | |
 |
| | Fig. 4 Schematic illustration showing the crystallite structure, phase transition and decomposition of γ-AlH3. | |
According to the heating rate and peak temperature of dehydriding (Tp) in the DSC curve, the Ea for the dehydriding reaction was obtained based on the slope measurement (see Fig. 5). The apparent activation energy for the dehydriding of γ-AlH3 and α-AlH3 in the nano-composite were estimated to be 66.8 kJ mol−1 and 74.7 kJ mol−1, which are slightly higher than that of these two AlH3 phases dopped with Zn and Zr (51.8 kJ mol−1 and 61.5 kJ mol−1), respectively. It is this decrease in kinetic barrier for dehydriding that contributes to the remarkable improvement in the dehydriding kinetics. This is believed to be the mechanism responsible for the faster dehydriding rate of the γ-AlH3/MgCl2 composite dopped with Zn and Zr, as shown in the aforementioned TPD and DSC results.
 |
| | Fig. 5 Determination of the apparent activation energy for dehydriding of γ-AlH3 and α-AlH3 by using Kissinger's equation and based on DSC measurements. The inset shows the Ea for dehydriding of γ-AlH3 and α-AlH3 dopped with Zn and Zr. | |
3.4 Microstructural evolution during dehydriding
SEM observation was carried out to characterize the morphological evolution of the γ-AlH3/MgCl2 nano-composite during dehydriding. Fig. 6 shows the SEM images of the nano-composite powders dehydrided at 240 °C for various times. The particle size of the nano-composite powders, as shown in Fig. 6(b), (d), and (f), was characterized based on the inserted graphs in the SEM images by using Image-ProPlus 6.0 software. As can be seen in Fig. 6(a) and (b), most particles after dehydriding at 240 °C for 2 h have a size of 0.5–1 μm, which is a little smaller than those of the as-prepared γ-AlH3/MgCl2 sample (∼1 μm),51 which suggests that the dehydriding reaction of the AlH3 phase leads to particle size refining of the nano-composite. With the increase of the dehydriding time to 4 h, both powder particle growth and agglomeration were observed, with the size of most particles being in the range of 1–3.5 μm, as shown in Fig. 6(c) and (d). For further extension of the dehydriding time to 6 h, severe agglomeration or aggregation, a phenomenon somewhat like sintering of the powder particles was observed, the morphology of the particles became irregular and difficult to identify, and the size of most particles was estimated to be in the range of 3.5–5 μm, as shown in Fig. 6(e) and (f). It was found by Graetz that the aggregation of particles will lead to the reduction of the specific surface area and the gateways for hydrogen diffusion.52 This can well explain the phenomenon observed in our previous work that the dehydriding kinetics of the γ-AlH3/MgCl2 composite declined with dehydriding time in the later stage of the isothermal dehydriding process.49,53
 |
| | Fig. 6 SEM images and particle size stats of γ-AlH3/MgCl2 nano-composite after isothermal dehydriding at 240 °C for various times: (a) and (b) 2 h, (c) and (d) 4 h, (e) and (f) 6 h. The insets show the images obtained by using Image-ProPlus 6.0. | |
Transmission electron microscopy (TEM) is a unique and effective way to characterize phase transitions in nano-structured materials. Although some hydrides were previously analyzed by TEM observation,54,55 so far the microstructure evolution related to the dehydriding process of the γ-AlH3/MgCl2 nano-composite has not been studied systematically. Fig. 7 shows the TEM images of the γ-AlH3/MgCl2 nano-composite dehydrided at different temperatures for 6 h. TEM observation confirmed that both direct decomposition of the γ-AlH3 phase in the nano-composite and its transition to α-AlH3 had occurred upon dehydriding at 90 °C for 6 h, as shown in Fig. 7(a)–(d). It can be seen from Fig. 7(a) that crystallites with average grain sizes of 50 nm and 5 nm were formed in the dehydrided sample, respectively. Moreover, two types of newly formed crystallites were highlighted using squares denoted by Y and Z to analyze phase composition. The selected area diffraction pattern (SADP) shown in Fig. 7(b) presents clear evidence that there exist α-AlH3, Al, MgCl2, and some retaining γ-AlH3 in the dehydrided sample. Shown in Fig. 7(c) and (d) are High Resolution Transmission Electron Microscopy (HRETM) images of the areas denoted by Y and Z in Fig. 7(a). As seen in Fig. 7(c), it is clear that the values of the identified lattice spacing, 3.23 and 2.22 Å, are in good accordance with the theoretical lattice distance of (012) and (110) planes of α phase, the lattice fringe with a spacing of 2.56 Å marked with a red arrow corresponds well to the (104) plane of MgCl2. Meanwhile, the parallel lattice fringe shown in Fig. 7(d) further identified that the relative smaller crystallite has a perfect match with the Al phase in the miller index. Namely, the lattice spacing of 2.02 Å between planes confirms the existence of Al. These observations indicated that a portion of γ-AlH3 decomposes directly, while the remaining γ-AlH3 transforms to α-AlH3. These observations have good agreement with above XRD analysis. After heating the γ-AlH3/MgCl2 nano-composite at 180 °C for 6 h, the bright field image revealed that much smaller crystallites emerged in the dehydrided product, as shown in Fig. 7(e). The HRTEM image obtained from part of these finer crystallites is shown in Fig. 7(f). These particles with much finer crystallites were identified to be the dehydriding product, i.e., Al/MgCl2 nano-composite. With the dehydriding temperature further increasing to 240 °C, severe agglomeration of the dehydriding product Al/MgCl2 composite was observed, as shown in Fig. 7(g). By comparing (g) with (e) in Fig. 7, the crystallite size of the Al/MgCl2 composite obtained by dehydriding at 240 °C was larger than when dehydrided at 180 °C for the same time. Moreover, it can be calculated from Fig. 7(g) that the average crystallite size of the composite increased rapidly to 45 nm. The HRTEM image shown in Fig. 7(h) indicates that values of the lattice spacing identified as 2.34, 2.02, and 1.98 Å are in good agreement with the lattice distance of Al (111), (200) and MgCl2 (107) planes, respectively.
 |
| | Fig. 7 TEM images of γ-AlH3/MgCl2 nano-composite after isothermal dehydriding at different temperature for 6 h: (a) 90 °C, (b) SADP of (a), (c) and (d) HRTEM of Y and Z in (a), (e) 180 °C, (f) HRTEM image of (e), (g) 240 °C, (h) HRTEM image of (g). | |
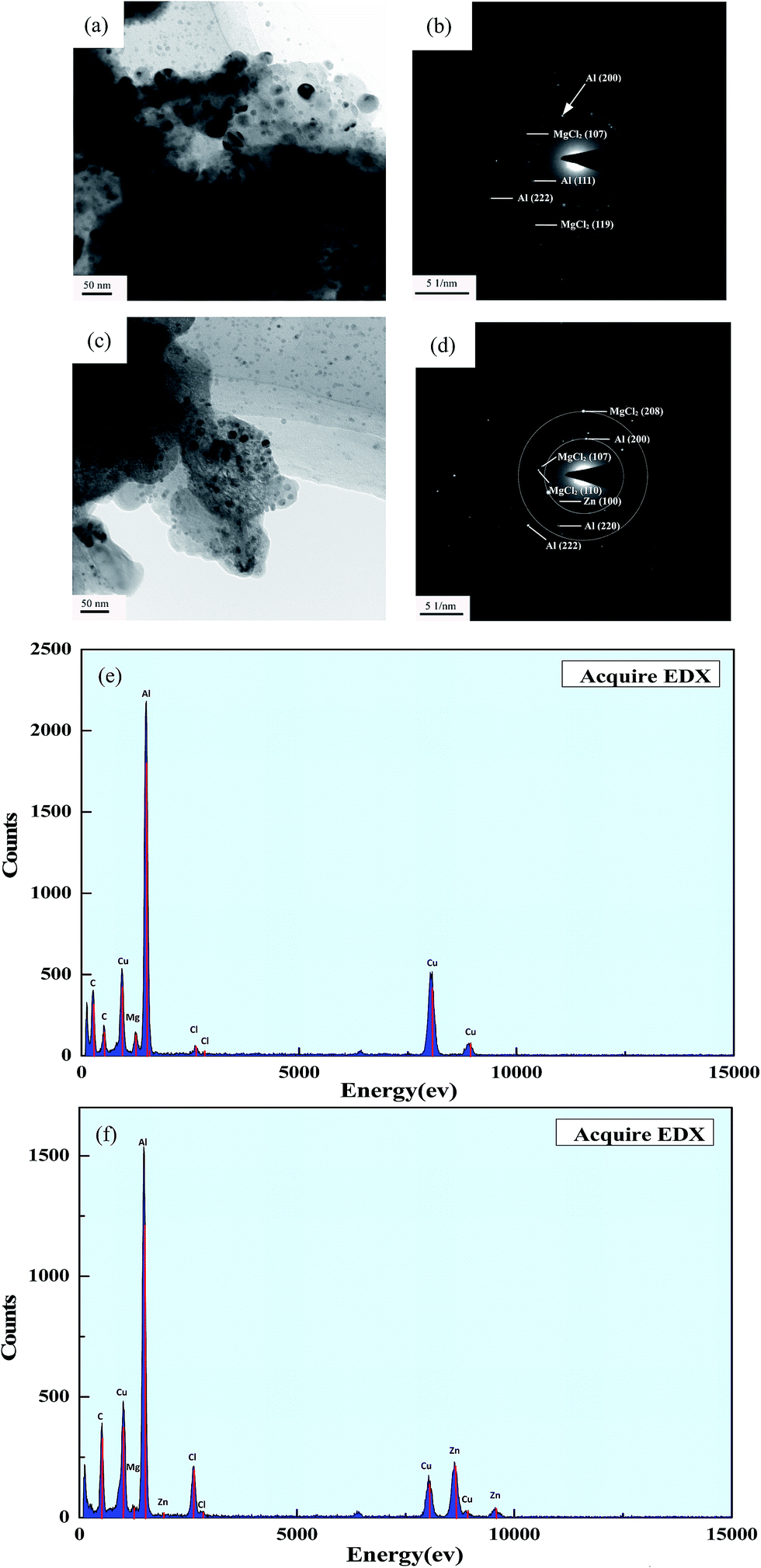
Based on the aforementioned TPD and DSC analysis, it is clear that, by dopping with Zn and Zr, the decomposition kinetics of γ-AlH3/MgCl2 nano-composite was accelerated. However, the underlying catalytic mechanism of Zn and Zr still needs to be clarified. To investigate the catalytic mechanism and the role that Zn and Zr play in the de-hydriding process, comparative TEM study of the as-dehydrided γ-AlH3/MgCl2 nano-composite samples with and without Zn and Zr dopping were performed, as shown in Fig. 8. It is seen from Fig. 8(a) that, after heating at 240 °C for 4 h, the average grain size of the as-dehydrided product, i.e., the Al/MgCl2 composite, was estimated to be around 35 nm. Moreover, the selected area electron diffraction patterns in Fig. 8(b) were identified to match well with the (119) and (107) planes of MgCl2 and (200), (111) and (222) planes of Al. Fig. 8(c) shows the TEM image of the Zn and Zr dopped nano-composite dehydrided at same condition as the sample without dopping. It is evident from Fig. 8(c) that AlH3 has transformed into Al and the average grain size of the Al/MgCl2 composite was estimated to be around 26 nm. The electron diffraction patterns in Fig. 8(d) were found to match well with the (100) plane of Zn and the (200), (220), (222) planes of Al as well as the (110), (208), (107) planes of MgCl2. This suggests that no intermetallic phase or intermediate hydride of Zn and Zr with AlH3 was formed during dehydriding. It was found that, by dopping with Zn and Zr, the temperature to initiate dehydriding of the nano-composite is reduced and the corresponding dehydriding rate accelerated. Similar effects on improving the dehydriding kinetic properties have been observed in other solid hydrogen storage materials such as NH3BH3.56 Song also demonstrated that Ti as a transition metal can efficiently increase the dehydrogenation rate of NaAlH4 by weakening the interaction between Al and H atoms.57 According to SEM observations in Fig. S1† and the aforementioned analysis, the role of the Zn and Zr dopping on the initial dehydriding period seems to be weakening the hydrogen bonding and preventing the AlH3 particles from agglomeration. Thus, by dopping with Zn and Zr, the starting temperature for hydrogen release can be lowered, and the dehydriding rate of the γ-AlH3/MgCl2 nano-composite is also faster. However, the rate of hydrogen release is also associated with a process involving nucleation and growth of Al. It was demonstrated by Liu that 3d transition metal and its hydride can act as an impediment to growth of metal Al and improve the dehydriding kinetics during the desorption process.58,59 Additionally, It was found by Graetz et al. that the dehydriding kinetic of AlH3 is controlled by nucleation and growth of Al in two and three dimensions.29 The larger the grain size of the formed Al, the lower the dehydriding rate of γ-AlH3/MgCl2 nano-composite was achieved during the dehydriding. Upon the above analysis, it can be deduced that the elements Zn and Zr have positive effects on the dehydriding kinetics of AlH3.
 |
| | Fig. 8 TEM images of nano-composite samples dehydrided at 240 °C for 4 h: (a) and (b) bright field image and corresponding ED pattern of γ-AlH3/MgCl2 nano-composite, (c) and (d) bright field image and corresponding ED pattern of γ-AlH3/MgCl2 nano-composite dopped with Zn and Zr, (e) and (f) EDS maps of nano-composite without and with Zn and Zr dopping. | |
4. Conclusions
The hydrogen desorption capacity of γ-AlH3/MgCl2 nano-composite synthesized by solid state reaction milling of MgH2 and AlCl3 as reagents achieves about 9.7 wt% when it is heated from 40 to 320 °C for non-isothermal dehydriding. By DSC analysis and TEM observation, the dehydriding mechanism was clarified. Firstly, at relatively lower temperature, though some γ-AlH3 in the nano-composite can decompose directly, most of this γ phase tends to transform to the thermodynamically more stable α-AlH3. Then, upon the subsequent dehydriding at higher temperature, both the remained γ-AlH3 and the newly formed α-AlH3 decompose directly, with the fast release of hydrogen and the formation of nano-sized Al crystallites, until the finish of the process. By using Kissinger's method, the activation energy for dehydriding of γ-AlH3 and α-AlH3 in the nano-composite was estimated to be 66.8 kJ mol−1 and 74.7 kJ mol−1, respectively. Zn and Zr dopping can not only reduce the activation energy for dehydriding of both the γ-AlH3 and the α-AlH3 (51.8 kJ mol−1 and 61.5 kJ mol−1), but also facilitate hydrogen diffusion by impeding the grain growth of the dehydriding product Al phase. Therefore, remarkable improvement in dehydriding kinetic properties is realized by Zn and Zr dopping.
Notes and references
- A. M. Seayad and D. M. Antonell, Adv. Mater., 2004, 16, 765–777 CrossRef CAS.
- U. Eberle, M. Felderhoff and F. Schueth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- G. Frenette and D. Forthoffer, Int. J. Hydrogen Energy, 2009, 34, 3578–3588 CrossRef CAS.
- M. Contestabile, G. J. Offer, R. Slade, F. Jaeger and M. Thoennes, Energy Environ. Sci., 2011, 4, 3754–3772 Search PubMed.
- Y. Wang, L. Li, F. Qiu, C. An, Y. Wang, L. Jiao and H. Yuan, J. Energy Chem., 2014, 23, 726–731 CrossRef.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- H. Liu, X. Wang, Z. Dong, G. Cao, Y. Liu, L. Chen and M. Yan, Int. J. Hydrogen Energy, 2013, 38, 10851–10856 CrossRef CAS.
- A. Züttel, Mater. Today, 2003, 6, 24–33 CrossRef.
- D. K. Ross, Vacuum, 2006, 80, 1084–1089 CrossRef CAS.
- P. Chen and M. Zhu, Mater. Today, 2008, 11, 36–43 CrossRef CAS.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- G. Sandrock, J. Reilly, J. Graetz, W. M. Zhou, J. Johnson and J. Wegrzyn, Appl. Phys. A, 2005, 80, 687–690 CrossRef CAS.
- J. P. Maehlen, V. A. Yartys, R. V. Denys, M. Fichtner, C. Frommen, B. M. Bulychev, P. Pattison, H. Emerich, Y. E. Filinchuk and D. Chernyshov, J. Alloys Compd., 2007, 446, 280–289 CrossRef.
- S. D. Beattie, T. Humphries, L. Weaver and G. S. McGrady, Chem. Commun., 2008, 4448–4450 RSC.
- K. Iyakutti, Y. Kawazoe, M. Rajarajeswari and V. J. Surya, Int. J. Hydrogen Energy, 2009, 34, 370–375 CrossRef CAS.
- D. Lacina, J. Wegrzyn, J. Reilly, Y. Celebi and J. Graetz, Energy Environ. Sci., 2010, 3, 1099–1105 CAS.
- E. M. Banach, H. A. Stil and H. Geerlings, J. Mater. Chem., 2012, 22, 324–327 RSC.
- F. M. Brower, N. E. Matzek, P. F. Reigler, H. W. Rinn, C. B. Roberts, D. L. Schmidt and K. Terada, J. Am. Chem. Soc., 1976, 98, 2450–2453 CrossRef CAS.
- J. Graetz, J. J. Reilly, J. G. Kulleck and R. C. Bowman, J. Alloys Compd., 2007, 446, 271–275 CrossRef.
- L. V. Dinh, D. A. Knight, M. Paskevicius, C. E. Buckley and R. Zidan, Appl. Phys. A, 2012, 107, 173–181 CrossRef CAS.
- P. Claudy, B. Bonnetot, J. Etienne and G. Turck, J. Therm. Anal., 1975, 8, 255–263 CrossRef CAS.
- B. Baranowski and M. Tkacz, Z. Phys. Chem., 1983, 135, 27–38 CrossRef CAS.
- J. Graetz and J. J. Reilly, J. Alloys Compd., 2006, 424, 262–265 CrossRef CAS.
- M. A. Petrie, J. C. Bottaro, R. J. Schmitt, P. E. Penwell and D. C. Bomberger, US Pat. 6 228 338, 2001.
- Y. Wang, G. K. Palsson, H. Raanaei and B. Hjorvarsson, J. Alloys Compd., 2008, 464, L13–L16 CrossRef CAS.
- P. B. Kempa, V. Thome and M. Herrmann, Part. Part. Syst. Charact., 2009, 26, 132–137 CrossRef CAS.
- S. Kato, M. Bielmann, K. Ikeda, S. I. Orimo, A. Borgschulte and A. Züttel, Appl. Phys. Lett., 2010, 96, 051912 CrossRef.
- J. Graetz and J. J. Reilly, J. Phys. Chem. B, 2005, 109, 22181–22185 CrossRef CAS PubMed.
- J. W. Turley and H. W. Rinn, Inorg. Chem., 1969, 8, 18–22 CrossRef CAS.
- V. A. Yartys, R. V. Denys, J. P. Maehlen, C. Frommen, M. Fichtner, B. M. Bulychev and H. Emerich, Inorg. Chem., 2007, 46, 1051–1055 CrossRef CAS PubMed.
- H. W. Brinks, C. Brown, C. M. Jensen, J. Graetz, J. J. Reilly and B. C. Hauback, J. Alloys Compd., 2007, 441, 364–367 CrossRef CAS.
- G. C. Sinke, L. C. Walker, F. L. Oetting and D. R. Stull, J. Chem. Phys., 1967, 47, 2759–2761 CrossRef CAS.
- S. Orimo, Y. Nakamori, T. Kato, C. Brown and C. M. Jensen, Appl. Phys. A, 2006, 83, 5–8 CrossRef CAS.
- B. Xu, J. Liu and X. Wang, Vacuum, 2014, 99, 127–134 CrossRef CAS.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS PubMed.
- J. Huot, G. Liang and R. Schultz, Appl. Phys. A: Mater. Sci. Process., 2001, 72, 187–195 CrossRef CAS.
- A. Zaluska, L. Zaluski and J. O. Ström-Olsen, Appl. Phys. A: Mater. Sci. Process., 2001, 72, 157–165 CrossRef CAS.
- J. Huot, J. F. Pelletier, L. B. Lurio, M. Sutton and R. Schulz, J. Alloys Compd., 2003, 348, 319–324 CrossRef CAS.
- G. Sandrock, J. Reilly, J. Graetz, W. M. Zhou, J. Johnson and J. Wegrzyn, J. Alloys Compd., 2006, 421, 185–189 CrossRef CAS.
- V. P. Balema, V. K. Pecharsky and K. W. Dennis, J. Alloys Compd., 2000, 313, 69–74 CrossRef CAS.
- Y. Kim, E. K. Lee, J. H. Shim, Y. W. Cho and K. B. Yoon, J. Alloys Compd., 2006, 422, 283–287 CrossRef CAS.
- J. A. Fernandez, F. Aguey-Zinsou, M. Elsaesser, X. Z. Ma, M. Dornheim, T. Klassen and R. Bormann, Int. J. Hydrogen Energy, 2007, 32, 1033–1040 CrossRef.
- S. Sartori, S. M. Opalka, O. M. Løvvik, M. N. Guzik, X. Tang and B. C. Hauback, J. Mater. Chem., 2008, 18, 2361–2370 RSC.
- M. Paskevicius, D. A. Sheppard and C. E. Buckley, J. Alloys Compd., 2009, 487, 370–376 CrossRef CAS.
- S. Sartori, A. Istad-Lem, H. W. Brinks and B. C. Hauback, Int. J. Hydrogen Energy, 2009, 34, 6350–6356 CrossRef CAS.
- J. Huot, D. B. Ravnsbæk, J. Zhang, F. Cuevas, M. Latroche and T. R. Jensen, Prog. Mater. Sci., 2013, 58, 30–75 CrossRef CAS.
- S. Gupta, T. Kobayashi, I. Z. Hlova, J. F. Goldston, M. Pruski and V. K. Pecharsky, Green Chem., 2014, 16, 4378–4388 RSC.
- C. W. Duan, L. X. Hu and D. Xue, Green Chem., 2015, 17, 3466–3474 RSC.
- C. W. Duan, L. X. Hu and Y. Sun, RSC Adv., 2015, 5, 17104–17108 RSC.
- C. W. Duan, L. X. Hu, Y. Sun, H. P. Zhou and Y. Huan, Dalton Trans., 2015, 44, 16251–16255 RSC.
- J. Graetz, J. J. Reilly, V. A. Yartys, J. P. Maehlen, B. M. Bulychev, V. E. Antonov, B. P. Tarasov and I. E. Gabis, J. Alloys Compd., 2011, 509, S517–S528 CrossRef CAS.
- C. W. Duan, L. X. Hu, Y. Sun, H. P. Zhou and Y. Huan, Phys. Chem. Chem. Phys., 2015, 17, 22152–22159 RSC.
- M. Felderhoff, K. Klementiev, W. Grünert, B. Spliethoff, B. Tesche, J. M. Bellosta von Colbe, B. Bogdanović, M. Härtel, A. Pommerin, F. Schüth and C. Weidenthalera, Phys. Chem. Chem. Phys., 2004, 6, 4369–4374 RSC.
- A. Léon, O. Kircher, H. Rösner, B. Décamps, E. Leroy, M. Fichtner and A. P. Guégan, J. Alloys Compd., 2006, 414, 190–203 CrossRef.
- D. J. Heldebrant, A. Karkamkar, N. J. Hess, M. Bowden, S. Rassat, F. Zheng, K. Rappe and T. Autrey, Chem. Mater., 2008, 20, 5332–5336 CrossRef CAS.
- Y. Song, J. H. Dai, C. G. Li and R. Yang, J. Phys. Chem. C, 2009, 113, 10215–10221 CAS.
- H. Liu, X. Wang, Y. Liu, Z. Dong, S. Li, H. Ge and M. Yan, J. Phys. Chem. C, 2014, 118, 18908–18916 CAS.
- H. Liu, X. Wang, Y. Liu, Z. Dong, G. Cao, S. Li and M. Yan, J. Mater. Chem. A, 2013, 1, 12527–12535 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reagents and procedure as well as SEM spectra, TPD, DSC curves are included in the ESI. See DOI: 10.1039/c6ra10491f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.