
Mini-review: fluorescence imaging in cancer cells using dye-doped nanoparticles
Ragini Jenkinsa,
Mary K. Burdettea and
Stephen H. Foulger*ab
aCenter for Optical Materials Science and Engineering Technologies, Department of Materials Science & Engineering, Clemson University, Clemson, SC 29634-0971, USA. E-mail: foulger@clemson.edu; Tel: +1-864-656-1045
bDepartment of Bioengineering, Clemson University, Clemson, SC 29634-0971, USA
First published on 27th June 2016
Abstract
Fluorescence imaging has gained increased attention over the past two decades as a viable means to detect a variety of cancers. Fluorescence imaging has the potential to provide physicians with high resolution images with enhanced contrast, which will allow them to be able to better diagnose and treat patients with cancer. Early detection and treatment are key to eradicating cancer in a patient, and fluorescence imaging has the ability to identify non-advanced, even pre-cancerous, tumors where imaging based on white light or radiation overlooked them. Several fluorescent dyes have been identified as possible fluorophores for enhanced fluorescence imaging, such as cyanine, squaraine, porphyrin, phthalocyanine, and borondipyrromethane dyes. These dyes have high fluorescence quantum yields, which provides a high target to background ratio; however, these dyes are often plagued by low water solubility. This low solubility can be ameliorated by conjugating or covalently attaching these dyes to polymeric crosslinked micelles, polymersomes, or polymer-core nanoparticles. These particle & dye systems then can become platforms on which secondary components can be attached to enhance the systems functionality. For example, dyes attached to these nanocarriers can target tumors through passive targeting; however, active targeting can be achieved by further modifying these nanocarriers with ligands that have a binding affinity for receptors overexpressed in tumor cells, cell surface receptors located on the tumor cell membrane, or endothelium. Fluorescence activation of the probes is another promising technology for the early detection of cancer. Activation requires that there be a change in fluorescence, whether it be an emission wavelength change or a fluorescence “on/off” signal when in the presence of some external stimuli. Activation increases the target to background ratio and enhances the contrast of the obtained image. This review serves to highlight the recent developments of (1) improved fluorescent dyes for the detection of cancer, with a specific focus on dyes that are being coupled to nanocarriers; (2) dye & nanocarrier systems that target, both actively and passively, tumors, and (3) fluorescence activation of these fluorophore systems for better image quality.
1 Introduction
Cancer, a disease caused by the uncontrollable growth of abnormal cells, is one of the leading causes of death worldwide, accounting for 13% of all deaths.1 Malignant cancer cells are more effectively treated when identified early in the disease. Identification of tumors using nanoparticle technologies is beginning to significantly impact molecular imaging methods.2 Molecular imaging plays a vital role in the healthcare sector, since abnormal conditions and diseases are often diagnosed through imaging, while therapeutic methods used for the treatment of the abnormalities are often guided by imaging. At present, there are various forms of imaging techniques employed including tomography, magnetic resonance imaging (MRI), gamma scintigraphy, ultrasound, and optical imaging (cf. Fig. 1). Computed tomography gives high resolution images and has the ability to differentiate between different tissues, but it has several drawbacks, such as high cost and the need of a contrast agent for an enhanced contrast, as well as exposing the tissue to radiation.3,4 Magnetic resonance imaging (MRI) does not require ionizing radiation and gives high resolution images with better contrast; however, this technique is expensive and has low sensitivity.3,5,6 Unfortunately, MRI cannot be used in patients with metal implants or metallic devices, such as pacemakers.7 Gamma scintigraphy, which includes positron emission tomography (PET) and single-photon emission computed tomography (SPECT), suffers from all three major drawbacks: ionizing radiation, low resolution, and high cost.7–9 There is a widespread use of ultrasound since its low cost, non-invasive, and an easily performed procedure with no radiation, but the images obtained are low in resolution.3,5,7 Optical imaging is a highly sensitive and low cost procedure7,10,11 that has the advantage that multiple probes with different spectral characteristics can be potentially employed for multichannel imaging.3,12,13 The setbacks for optical imaging are that the images obtained are generally low in resolution and have a limited tissue penetration7 to light in the UV-vis regime. Tissues exhibit a reduced absorption to light in the far red (i.e. 775–925 nm) out to the near-infrared (NIR) range14 and this review examines recent developments in the field of molecular imaging with fluorophores that emit in these regions. | ||
| Fig. 1 Standard molecular imaging instruments: (a) magnetic resonance imaging (MRI); (b) computed tomography (CT); (c) positron emission tomography (PET); (d) single-photon emission computed tomography (SPECT); and (e) optical imaging (f) ultrasound (reprinted with permission from ref. 7 Copyright 2010 Elsevier B. V.). | ||
The use of NIR fluorophores in medical imaging has been gaining popularity. These fluorophores overcome some drawbacks present with visible light emitting dyes, such as background noise, and also facilitate deeper imaging in the human body.15 Unfortunately, small molecule fluorophores have four major limitations: (1) limited aqueous solubility, (2) short in vivo circulation lifetimes, (3) low quantum efficiencies, and (4) low signal to noise ratios. These drawbacks can be ameliorated with varying success by either encapsulating the fluorophore inside a nanoparticle or attaching it on the surface of the nanoparticle. Over the years, fluorescent nanocarriers such as surface cross-linked micelles,16,17 dendrimers,18 biodegradable polymeric nanoparticles,19,20 magnetic and other metal particles,21,22 and liposomes21 for tumor diagnosis have been developed. Among these technologies, platforms based on polymeric materials, especially biodegradable nanoparticles, are of particular interest due to the flexibility offered by macromolecular synthesis methods, high drug loading capacities, improved drug solubility, and their ease of multifunctionalization.23–25 In this review, the developments in fluorescence imaging with the three polymer based platforms: cross-linked micelles, polymersomes and polymer-core nanoparticles (cf. Fig. 2), when coupled with the commonly used fluorophores, will be explored.
 | ||
| Fig. 2 Schematic structures of (a) polymer cross-linked micelles, (b) polymersomes, and (c) polymer-core nanoparticles (adapted with permission from ref. 26 Copyright 2012 Multidisciplinary Digital Publishing Institute AG). | ||
2 Small molecule imaging
Small molecule organic fluorophores have been in demand in the biomedical community for imaging and image guided surgery or therapy.27–34 Imaging with far-red and near-infrared absorbing and emitting dyes such as cyanine, squaraine, thiazine, oxazine, porphyrins, and phthalocyanines, is commonplace and most of them have been approved for clinical trials.35–47 Recently, a new class of dyes, borondipyrromethane (BODIPY) derivatives, have emerged and gained popularity.48–53 However, imaging with small molecules have some challenges in both their optical performance and delivery to the tissue of interest, such as water insolubility, aggregation, low quantum yield, insufficient photostability, low tumor to background ratio, and short in vivo circulation lifetimes. Researchers continue to focus on improving these dyes to enhance their light output and localization in specified regions and are detailed in the following section.2.1 Cyanine dyes
The general structure of cyanine dyes consists of two aromatic nitrogen-containing heterocycles, acting as both electron donors and acceptors. The two aromatic rings are connected by an odd number of methine groups in which (n + 1) bi-electrons are distributed over n atoms that produce a delocalized cation across the methine chain (cf. Fig. 3a).54–56 Depending on the methine chain length, the cyanine dyes absorb in the visible to infrared regions of the electromagnetic spectrum.57 In 1856, C. H. G. Williams synthesized the first cyanine dye57,58 and since this time many analogs with varying lengths of the methine chain have been developed and employed in various biomedical applications ranging from angiography to photodynamic therapy.54 The majority of commercial fluorescent probes for in vivo imaging consist of cyanine dyes. Of all the cyanine dyes, indocyanine green (ICG) is the most popular and was approved by the FDA decades ago for evaluating blood flow and clearance.54,55 ICG and many other dyes generally exhibit high molar extinction coefficients but low quantum yields, poor photostability, high plasma protein binding rate, and undesired aggregation.59 To overcome some of the limitations, new analogs that have a cyclohexenyl in the middle of the methine chain and moieties such as carboxylic and sulfonate groups were introduced.60,61 These alterations result in the improved water solubility of the dye, photostability, and quantum yield. | ||
| Fig. 3 Structures of (a) cyanine, (b) squaraine, (c) phthalocyanine, and (d) boron dipyrromethane dyes. | ||
2.2 Squaraine dyes
Squaraine dyes are 1,3-zwitter ionic donor–acceptor–donor (D–A–D) structures with the central acceptor squaryl ring containing donor aromatic or heterocyclic rings on each side (cf. Fig. 3b).62 In 1965, Treibs and colleagues first reported the synthesis of squaraine dyes, which have extremely intense absorption bands, high molar coefficients, and good photoconductivity.54,63,64 Despite these molecules having excellent physical–chemical properties, they gained importance only in the late 2000s due to their limitations such as low water solubility, propensity for aggregation, and poor chemical stability with only a few analogs of the dye emitting at wavelengths higher than 800 nm.54 Most importantly, the squarate bridge is susceptible to chemical attack by nucleophiles due to its electron deficiency and results in loss of fluorescence.65,66 In 2005, Gassensmith and colleagues synthesized a rotaxane molecule encapsulating a squaraine dye. The rotaxane cage protected the squaraine ring and inhibited aggregation, which improved the chemical and photostability of the dye.67 In order to further protect the squarate bridge from nucleophilic attack, a squarine that is dicyanovinyl functionalized was synthesized.65 In 2007, Umezawa and colleagues developed another alternative squaraine derivative dye with improved hydrophilicity due to four water-solubilizing sulfonate moieties added to the general structure.68 Squaraine dyes can be relatively challenging to synthesize, but it was shown that a microwave can actually be used to assist in the synthesis of 2,3,3-trimethylindolenine-based squarine dyes exhibiting maximum absorbance between 625 nm and 700 nm and emissions between 635 nm and 800 nm. These microwave synthesized dyes exhibited a better yield and reduced reaction time when compared squariane dyes synthesized the conventional way. In addition, it was shown that both symmetric and nonsymmetric dyes could be easily synthesized with this method.69 All these improvements to squaraine dyes have made them a very promising candidate for protein detection and in vivo imaging.2.3 Porphyrin and phthalocyanine dyes
Porphyrins are tetrapyrrolic molecules that consist of four pyrrolic sub-units linked on opposing sides through four methine (CH) bridges. Phthalocyanine molecules are an extension of porphyrins where each pyrrolic ring of porphyrin is extended by a benzene ring (cf. Fig. 3c).70 The central cavity of the phthalocyanine contains two hydrogen atoms and these atoms can be replaced by more than 70 different types of metal atoms.71–73 In addition, a variety of substituents can be added to the periphery of the macrocycle or to the axial positions of the central atom.54,74 These blue or green colored dyes absorb strongly in the red and near-infrared part of the visible spectrum. In 1907, Braun and Tcherniac synthesized the first metal-free and copper phthalocyanines.75 In vitro fluorescence imaging with phthalocyanines and porphyrins dates back to late the 1980s and early 1990s.76 In 1993, photofrin, a porphryin derivative, was the first chromophore in this class of dyes to be approved for clinical use for the treatment of bladder cancer.70 Since then there have been several analogs of the porphyrin and phthalocyanine synthesized and utilized.77–81 The low water solubility exhibited of these dyes has decreased their popularity for imaging.80,82–84 However, a new phthalocyanine chemistry was used to create a series of PEGylated cationic molecules, which has promising in vitro behavior against cancer cells.85 Click reactions have been employed to create phthalocyanines with different architectures; 4-arm star polymers utilizing either polystyrene or poly(tert-butyl acrylate) were synthesized with symmetrically tetra terminal alkynyl-substituted phthalocyanines as the core. The central metal atom in these systems was either copper or zinc.86 This type of architecture could be used to improve water solubility and prevent aggregation of the phthalocyanines due to the extension of the polymeric arms which disrupts the π stacking of the phthalocyanine. In 2015, Bandera and colleagues synthesized novel silicon phthalocyanine derivatives equipped with azide or alkyne functionality to be able to use the dyes in “click” chemistry, which allows the dyes to be used in an engineered nanodevice for cancer theranostics.87 In 2013, a water soluble porphyrin (THPP), with zinc metal as the central atom was developed and it showed an increase in in vitro photodynamic therapy activity by 2–3 times when compared to photofrin.88 Photodynamic therapy relies on the ability of the fluorophore, when excited at the appropriate wavelength of light, to generate a reactive oxygen species (singlet oxygen) from molecular oxygen through an intersystem crossing.89 In the field of tumor theranostics, these dyes have been more successful than the other classes of dyes due to their robust fluorescence, which yields images with better contrast and signal to noise ratios, as well as lending themselves to superior performance in photodynamic therapy. Yumita and colleagues synthesized a novel porphyrin derivative and demonstrated a mechanism that is related to the generation of singlet oxygen to achieve destruction of cancer cells through sound rather than light.902.4 Borondipyrromethane dyes
Borondipyrromethanes (BODIPY) have a general structure of 4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene (cf. Fig. 3d). In 1968, Treibs and Kreuzer first reported the synthesis of BODIPY dyes.91 These molecules typically have the following characteristics: sharp fluorescence with high quantum yield, excellent thermal and photochemical stability, and extinction coefficients around 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.92 Despite having such attractive properties, BODIPYs were not preferred because they emit in the yellow to deep-red region, and the extinction coefficients were considered to be relatively low.54 To overcome these disadvantages, two approachs have been undertaken. First, polymeric and copolymeric BODIPY dyes were synthesized to shift emission to the near-infrared region.93 Second, modification of the pyrrole core shifts the emission to the red end of the visible spectrum. The most significant modification is to include aryl groups to the BODIPY. Using this modification, BODIPYs with molar extinction coefficients of 253000 M−1 cm−1 and emission in the near-infrared region can be obtained.94,95 In 2014, BODIPY derivatives were synthesized via a Suzuki reaction where a hetero aryl substituent on the 3- or 3,5-position resulted in approximately a 150 nm bathochromic shift in both maximum absorbance and maximum emission. This series of BODIPYs resulted in an improved production of singlet oxygen.96 An increased fluorescence quantum yield (approximately 0.82) was achieved through the modification of the phenyl group with 2 methyl substituents in the meso position. These methyl groups provided rigidity to the molecule and resulted in a red shift in maximum emission to 644 nm (in chloroform) or 649 (in DMSO). These BODIPYs were further modified by a terminal bromo group in order to attach the dye onto carbon nano onions.97 This modification showed that BODIPY could be modified in a variety of ways in order to attach the dye to a scaffold, which could serve to incorporate them into a system for possible biomedical applications. In 2016, Kim and colleagues synthesized new BODIPY fluorophores that were capable of 1 or 2 photon fluorescence imaging. Further, they showed that the free fluorophores accumulate in the lysosomes of cells. The fluorophores have low cytotoxicity, which makes them ideal for imaging agents.98
000 M−1 cm−1.92 Despite having such attractive properties, BODIPYs were not preferred because they emit in the yellow to deep-red region, and the extinction coefficients were considered to be relatively low.54 To overcome these disadvantages, two approachs have been undertaken. First, polymeric and copolymeric BODIPY dyes were synthesized to shift emission to the near-infrared region.93 Second, modification of the pyrrole core shifts the emission to the red end of the visible spectrum. The most significant modification is to include aryl groups to the BODIPY. Using this modification, BODIPYs with molar extinction coefficients of 253000 M−1 cm−1 and emission in the near-infrared region can be obtained.94,95 In 2014, BODIPY derivatives were synthesized via a Suzuki reaction where a hetero aryl substituent on the 3- or 3,5-position resulted in approximately a 150 nm bathochromic shift in both maximum absorbance and maximum emission. This series of BODIPYs resulted in an improved production of singlet oxygen.96 An increased fluorescence quantum yield (approximately 0.82) was achieved through the modification of the phenyl group with 2 methyl substituents in the meso position. These methyl groups provided rigidity to the molecule and resulted in a red shift in maximum emission to 644 nm (in chloroform) or 649 (in DMSO). These BODIPYs were further modified by a terminal bromo group in order to attach the dye onto carbon nano onions.97 This modification showed that BODIPY could be modified in a variety of ways in order to attach the dye to a scaffold, which could serve to incorporate them into a system for possible biomedical applications. In 2016, Kim and colleagues synthesized new BODIPY fluorophores that were capable of 1 or 2 photon fluorescence imaging. Further, they showed that the free fluorophores accumulate in the lysosomes of cells. The fluorophores have low cytotoxicity, which makes them ideal for imaging agents.98
3 Imaging with polymer based nanoparticles
It is well established that nanosized objects accumulate more efficiently in tumors and increase the target to background ratio due to the enhanced permeability and retention (EPR) effect.99 The EPR effect results in passive targeting of nanoparticles to tumor sites. Blood vessels supplying tumor tissues have larger pore sizes compared to those in healthy tissue, and tumor tissues have poor lymphatic drainage. These factors combined allow for a preferential tumor accumulation of nanoparticles.100–103 Small animal studies indicate that a 50 fold increase in accumulation of nanoparticles within tumor tissues are due to the EPR effect, coupled with an increase in in vivo circulation lifetimes.103 Apart from the increased accumulation and lifetimes, the nanoparticles have another advantage: they provide a platform for an increased surface area per volume for enhanced loading of imaging and therapeutic moieties.102 To this end, conjugating a small molecule fluorophore to the surface of the nanoparticle is preferred over using the fluorophore by itself for optical imaging. In addition to the EPR effect, the particle characteristics such as size distribution, surface charge, biocompatibility, biodegradation behavior, and availability of functional groups for conjugation play an important role in determining the circulation lifetimes and accumulation behavior in tumor tissues.26,102 It is established that biodegradable microparticles made of starch, albumin, or poly lactic acid, are rapidly cleared by the reticuloendothelial system (RES) and are unable to enter capillaries, making them less attractive for imaging applications.102 As mentioned previously, polymeric nanocarriers are of particular interest due to the flexibility offered by macromolecular synthesis methods, high drug loading capacities, improved drug solubility, and ease of multifunctionalization.23–25 The first preparation and characterization of polymeric nanoparticles was reported in 1976; since, the research in this area has grown exponentially.102,104 It has been established that neutrally charged particles, with an average diameter of 10–100 nm, and molecular weights up to 800 kDa were required to obtain longer circulation lifetimes.102,105 In addition, the choice of the parent polymer is important in designing such systems because the polymer tends to determine the final behavior of the system in various environments.102 Based on the above mentioned requirements for the nanocarrier, some of the successful organic fluorescent carriers used in fluorescence imaging will be discussed in detail in the following sections (cf. Fig. 2).3.1 Crosslinked micelles
A crosslinked micelle (CM) is a nanocarrier that is based on a self-assembled aggregation of amphiphilic molecules or surfactants with the fluorescent probes conjugated to it. Micelles have a dynamic structure in equilibrium and, at a critical micellar concentration (CMC), they exist as free monomers, so care should be taken to ensure that the concentration is below the CMC to avoid a fast and undesirable release of the probe upon administration.26 Typically, CMs are in the 5–150 nm size range,106 which allows extravasation and permeation into tissues and avoids clearance by the RES.107 The CMs are simple aqueous based preparations with highly tunable emission wavelengths and an expanded Stokes shift of the dye from 20 nm to about 160 nm,108 which made them an attractive carrier for fluorescence imaging.109 In addition, most CMs are not toxic and are biologically compatible. The exception is surfactant based micelles as the large amounts of surfactant used in their formation might lyse the cell membrane and denature proteins, so they are not a preferred design.107 In polymer based micelles, the hydrophobic units of the block copolymers form the hydrophobic core of the micelle and hydrophilic units surround the core forming a hydrophilic shell, thus making the micelle sterically stable and protecting it from EPS uptake.106,107 Using this strategy, a number of block co-polymer micelles that encapsulate near infrared emitting chromophores have been synthesized over the years and utilized in imaging applications.26,110,111 Recently, in 2013, Chen and colleagues synthesized PEGylated tripropargyl amine micelles conjugated to BODIPY fluorophores, which had excellent water solubility and membrane permeability. These compounds exhibited an increased Stokes shift without any covalent structure modification of the fluorophore.108,112 In addition, these BODIPY conjugated PEGylated tripropargyl amine micelles were observed to readily penetrate cell membranes and preferentially accumulate in the cytoplasm rather than the nucleus of the cell (cf. Fig. 4).112 In most of the micelles, poly(ethylene glycol) (PEG) remained as the choice of the hydrophilic unit as this polymer is widely accepted as a biocompatible material and is easily available.106 The recent advances in crosslinked micelles have come in the form of “smart” micelles that respond to various biological stimuli, including the pH of the environment, and can be designed to target specific tissues (cf. Fig. 5).106,113 Apart from synthesizing stimuli responsive micelles, there has been development of micelles based on recombinant proteins. Kim and colleagues114 developed a 50 nm recombinant protein micelle for targeted in vivo imaging. In these micelles, multiple fluorescent moieties were covalently conjugated to surface amines of crosslinked amphiphilic elastic-mimetic protein micelles utilizing common protein chemistry, namely N-hydroxysuccinimide ester chemistry. These micelles offered enhanced biocompatibility and can be further modified in order to tailor the micelle nanocarrier for specific applications. Self assembled micelles consisting of poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate) (PEG-PCC) covalently attached to amine modified indocyanine green (NH2-ICG) were successfully produced and showed efficient photodynamic therapy behavior (i.e. significant singlet oxygen generation).115 | ||
| Fig. 4 (a) Fluorescence, (b) transmission, and (c) overlapping fluorescence & transmission images of HeLa cells observed with confocal laser scanning microscopy. The cells were incubated for 30 min with 1 mM of BODIPY conjugated surface crosslinked micelles (ca. 10 nm) at 37 °C in an atmosphere of 95% air and 5% CO2. Fluorescence signals were detected between 570 nm and 620 nm with a constant excitation of 561 nm (reprinted with permission from ref. 108 Copyright 2013 The Royal Society of Chemistry). | ||
 | ||
| Fig. 5 Temperature induced self-assembly of ELPBC to form spherical micelles with multivalency. An N-terminal ELP[V1A8G7−n] gene (hydrophilic, high Tt) and C-terminal ELP[V5−n] gene (hydrophobic, low Tt) are fused together to create a gene that encodes an ELPBC. When the size and ratio of the blocks are optimal, the ELPBC self-assembles into a spherical micelle at approximately 40 °C. In the schematic, upon self-assembly the spherical micelles present several copies of an affinity targeting moiety (green triangle) and capture a drug or imaging agent (lightning bolt) within the core of the micelle (reprinted with permission from ref. 113 Copyright 2008 American Chemical Society). | ||
3.2 Polymersomes
Polymersomes are artificial vesicles or small hollow spheres that enclose a solution and have radii ranging from 50 nm to 50 μm.116 They have a large hydrophilic reservoir and a thick hydrophobic lamellar membrane that supports the storage of a large quantity (i.e. more than 10 mol/wt%) of hydrophobic fluorescent dyes and can protect the dyes from quenching and degrading.117,118 The polymer chains dictate the average fluorophore–fluorophore interspatial separation along with the fluorophore-localized electronic environment.116 Greater fluorescence was obtained for more hydrophobic fluorophores when relatively apolar membranes, such as poly(γ-methyl-ε-caprolactone), were used, and the more amphiphilic fluorophores were better dissolved when bilayers of poly(ε-caprolactone) were employed.118 For most polymersomes, the outer shell is a layer of dense polyethylene oxide (PEO), which confers the “stealth” like character and results in increased biocompatibility, structural integrity in plasma, and circulation lifetimes.116,117,119 In addition, the surface has terminal functional groups, such as alcohols, which can be used to covalently conjugate targeting molecules, drugs, and biomolecules to create a multifunctional unit.117 There has been extensive work and investigation into developing polymersomes into stimuli responsive multimodality agents,120–123 but for this review, we will focus on the polymersomes that contributed significantly for fluorescence imaging.118,119,124 Initially, Hammer and colleagues synthesized poly(ethylene oxide)-block-poly(ethylethylene) (PEO-b-PEE) diblock copolymers based polymersomes and expanded that technique to generate a number of biocompatible PEO-based amphiphilic block copolymers, such as poly(ethylene oxide)-block-poly(butadiene) (PEO-b-PBD).119 They loaded these polymersomes with hydrophobic dyes, such as Nile Red, and hydrophilic dyes, such as calcein, and demonstrated its use in deep-tissue fluorescence based imaging.119,125 PEO-b-PBD polymersomes encapsulating porphyrin based fluorophores generated a fluorescence signal that was penetrable through 1 cm of solid tumor.119 However, these systems were not biodegradable and were not fully biocompatible. To this end, Ghoroghchian and colleagues developed the first set of self-assembled polymersomes that were composed entirely of United States Food and Drug Administration (FDA) approved biodegradable diblock copolymers. They were fully bioresorbable diblock copolymers of poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL) and the diblock copolymer poly(ethylene oxide)-block-poly(γ-methyl ε-caprolactone) (PEO-b-PMCL) (cf. Fig. 6).118 In 2012, Massignani and colleagues developed poly(2-(methacryloyloxy)ethyl phosphorylcholine)-poly(2-diisopropylaminoethyl methacrylate) (PMPC-PDPA) diblock copolymers based polymersomes and loaded it with rhodamine, which had enhanced emission compared to free rhodamine and CellTracker dye.126 This increase was attributed to the effective dye delivery when the polymersome was used. In 2014, Quan and colleagues synthesized a fluorescent polymersome that could encapsulate hydrophobic or hydrophilic drugs instead of the traditional polymersome encapsulating fluorophores. It was prepared by self-assembly of block copolymer hydrophilic poly(ethylene glycol) borondipyrromethenes (MPEG-BODIPY) in aqueous solution. These ca. 57 nm particles exhibited low toxicity and high accumulation in tumor site through passive targeting.127 Polymersomes are also being used to encapsulate superparamagnetic iron oxide nanoparticles (SPIONs), an ubiquitous imaging agent in MRI. These polymersomes consist of either zwitterionic or cationic block copolymers (poly(2-methylacryloyloxy)ethyl phosphorylcholine)-block-((3-(methacryloylamino(propyl)trimethylammonium chloride)); PMPC20-b-PMAPTAC190) or zwitterionic and anionic block copolymers (poly(2-methylacryloyloxy)ethyl phosphorylcholine)-block-poly(sodium 2-(acrylamido)-2-methylpropanesulfonate; PMPC20-b-PAMPS196). In order to better encapsulate SPION, the SPIONs were coated with a cationic chitosan derivative that had been further modified with the fluorophore Alexa Fluor® 647. The fluorophore provides additional imaging capabilities and tracking. These polymersomes have a semipermeable membrane that is capable of encapsulating a large amount of highly charged nanoparticles; the chitosan coated SPIONs reside in the interior and in the membrane of the polymersome. Through laser scanning confocal imaging of the Alexa fluorophore, it was seen that the particles are uptaken by the endothelium.128 Polymersomes have started to be produced from chemically modified proteins in conjunction with polymers. Polymersomes of a poly ion complex based on chemically altered chitosan were used in conjunction with fluorescent quantum dots (QDs), where the hydrophobic QDs were encapsulated by the polymersome. The in vivo study showed that there was a longer retention time and greater fluorescence signal in the tumor for the polymersomes conjugated with QDs when compared to the isolated QDs. The fluorescence signal of the free QDs was quickly extinguished within 1 hour after treatment with a strong fluorescence signal in the liver suggesting that the free QDs are rapidly cleared from the tumor.129 In another study, poly(N-isopropylacrylamide) (PNIPAM), a thermally responsive polymer, along with a protein, namely a derivative of green fluorescent protein (amilFP497), was developed. The polymersome is a block copolymer with PNIPAM-b-amilFP497. These polymersomes exploit the conformation changes that occur when PNIPAM is subjected to temperatures in the lower critical solution temperature (LCST) of 37 °C. At temperatures below 37 °C, the polymersomes self assemble and are capable of encapsulating drugs, such as doxorubicin (DOX), and fluorophores during the self assembly process. The fluorophore phycoerythrin 545 was used in this study and was found to be in the core of the polymersome, while DOX was found in both the core and the membrane of the polymersome. At temperatures above the LCST, the polymersome unfolds and releases its load.130 | ||
| Fig. 6 Cartoon representing various means of incorporating oligo(porphyrin)-based NIRFs within polymersomes. (a) The NIRFs are different as they have various numbers of porphyrin subunits (N), various linkage topologies between porphyrin molecules, and various positions of ancillary aryl-group substituents (R), and the nature of each R group is different based on its chemistry. (b) Diblock copolymer at a variety of compositions have been used to generate polymersomes that have emission in the NIR. (c) Interfacial interactions between polymers and ancillary aryl group substituents, and the variety of conformational populations in which the NIRF can arrange itself, are valuable tools when manipulating and/or tuning the emission of the NIRF. (d) Control over the chemical composition and polymersome membrane thickness forces individual NIRFs into dielectric environments of matching polarity. (e) A family of nanoscale polymersomes possessing emission wavelengths corresponding to the NIR region of the visible spectrum (reprinted with permission from ref. 118 Copyright 2009 John Wiley & Sons Inc.). | ||
3.3 Polymer-core and fluorophore nanoparticles
The polymer-core nanoparticles can be divided into two categories: natural and synthetic. As the name of the category suggests, particles made from naturally occurring polymers such as dextran, albumin, gelatin, chitosan, heparin, bacteriophages, or lipoproteins fall under the natural category, and the particles synthesized from artificially produced polymers such as poly(amino acids), poly(alkyl-cyano acrylates), poly(esters), poly(orthoesters), poly(urethanes), and poly(acrylamides) are considered synthetic.26,102 In this current review, the focus is on synthetic polymer-core nanoparticles. The polymer-core nanoparticles have an inner core made of a cross-linked polymer while the fluorophore is either encapsulated within the core or covalently conjugated to the polymer. Encapsulation protects the fluorophore from direct interactions that could decrease its emission, but, at the same time, the fluorophore can neither form beneficial host/guest assemblies nor complex with biomacromolecules. Physical encapsulation of the fluorophore further suffers from leakage and non-specific release of the fluorophore. These drawbacks are overcome easily when the fluorophore is covalently conjugated to the outside of the particle.26,131 For the inner core, hydrophobic polymers such as poly(lactic-co-glycolic acid) (PLGA), poly(3-caprolactone) (PCL) and poly(methyl methacrylate) (PMMA) are preferred, as the hydrophobic matrix provides a strong affinity for the entrapment of poorly water solubilized fluorophores and drugs.132,133 Typical nanoparticle size varies from 50–500 nm, and the optimum nanoparticle size for tumor accumulation is between 70 and 200 nm as the nanoparticle uptake by the RES is in the size range between 150 and 300 nm.134,135 In addition, it has been well established that a coating of PEG on the hydrophobic particle is necessary to render the nanoparticles biocompatible, avoid rapid clearance by the RES, and increase the circulation lifetime, which increases accumulation and allows distribution in different tissues.26,131,136–138As mentioned previously poly(lactic acid) (PLA) based systems are the most popular choice for nanoparticles. PLA and its copolymers are one of the most extensively investigated matrix materials for nanoparticle-based diagnostic and drug delivery applications as they offer excellent biocompatibility, biodegradability into metabolizable moieties, and manufacturability.132,139 In addition, PLGA is approved by the FDA and is a component of many biodegradable market products for parenteral application.133,140 Initially, in 2004, Saxena and colleagues prepared 300–410 nm PLGA nanoparticles entrapping ICG by a modified spontaneous emulsification solvent diffusion method and were able to achieve over 74% loading of the fluorophore in the particles.141,142 Later in 2006, they demonstrated the use of these particles for tumor diagnosis and photodynamic therapy and also determined the biodistribution of ICG loaded PLGA nanoparticles in healthy C57BL/6 mice (female, 10 week old).143 In 2011, Schadlich and colleagues encapsulated different Nile Red and DiR (a carbocyanine based fluorophore) in PEGylated PLA nanoparticles and performed in vivo and ex vivo imaging studies to investigate the impact of particle size in tumor accumulation, distribution, and elimination.133,144 In addition to encapsulated systems, there are covalently conjugated fluorophore–PLA or PLGA nanoparticle systems being investigated. In 2010, Tong and colleagues developed Cy5-conjugated polylactide (Cy5-PLA) nanoparticles. They found these particles to have excellent signals with high tumor to background ratio fluorescence in various organs when administered intravenously to balb/c mice (cf. Fig. 7).145 In 2012, Reul and colleagues, evaluated the concept of covalently attaching a NIR dye to poly(lactide-co-glycolide) (PLGA) to create stable NIR fluorescent nanoparticles. For their studies, they coupled PLGA with DY-700 (a near infrared fluorophore) and injected the system intravenously into mice. According to their results, these particles had very good stability and preferentially accumulated in the liver where the free fluorophore would not enter. This system could be developed further into a finely traceable PLGA nanosystem for fluorescence imaging.140
 | ||
| Fig. 7 In vivo biodistribution of intravenously administered Cy5-PLA nanoparticles. Lateral tail vein injection using the LI-COR Odyssey scanner was performed and the biodistribution of Cy5-PLA nanoparticles was obtained. More than a 10-fold increase in accumulation was seen in Cy5-PLA nanoparticles contained in visceral organs when compared to background autofluorescence. Cy5-PLA nanoparticles showed the most accumulation in the spleen when compared to the other organs sampled (i.e. spleen, liver, kidney, lung, and heart); bar scale is 5.5 mm (reprinted with permission from ref. 145 Copyright 2010 WILEY-LISS INC.). | ||
Apart from PLA based nanosystems, there have been particles based on other polymers such as poly(propargyl acrylate) (PA), poly(styrene-co-methacrylic acid), and poly(acrylic acid) (PAA). In 2011 Rungta and colleagues prepared sub-100 nm PA nanoparticles which had azide-terminated indocyanine green (azICG) covalently conjugated to the surface. The azICG was attached to the particles via azide/alkyne Huisgen cycloaddition using a copper catalyst.59 Since the inception of the “click” cycloaddition reaction in the field of drug discovery in 2001 by Sharpless and coworkers, it has been used by numerous researchers to prepare nanosized particles including polymeric micelles and nanoparticles, liposomes and polymersomes, capsules, metal and silica nanoparticles, carbon nanotubes and fullerenes, and bionanoparticles for tumor diagnostic and drug delivery applications.146–160 In addition, significant growth and interest is seen in developing new processes for the “click” reaction to achieve high reaction kinetics, excellent biocompatibility, selective labeling of specific targets, easily accessible reactive tags, and long shelf-life.146 In the future, it can be expected that a majority of the nanosystems for tumor theranostics will utilize the “click” reaction.161,162 These PA-azICG nanoparticles particles demonstrated great potential for photodynamic therapy (PDT), but their brightness was too low for imaging applications. Later in 2013, the same group synthesized particles that were surface modified with an azide terminated squaraine dye (azSQ) instead of azICG; these PA-azSQ particles performed better than the PA-azICG particles for fluorescence imaging. Co-localization studies were performed to establish that the uptake of the PA-azSQ particles was through endocytosis and in vitro fluorescence imaging was carried out in UMSCC22A (head and neck) and A549 (lung) cancer cells.131 In 2011, Palma and colleagues, prepared BF2 chelated tetraarylazadipyrromethene (near infrared dye) conjugated poly(styrene-co-methacrylic acid) particles. The dye was attached to the nanoparticles via a standard N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) coupling reaction. Using those particles, they were able to obtain promising results for real time in vitro imaging in MDAMB-231 (breast cancer), HEK293T (kidney), and CAKI-1 (renal cancer) cell lines (cf. Fig. 8).163 In 2013, Tao and colleagues reported the synthesis of the first biocompatible NIR-II agent nanoparticles, for in vivo imaging. These particles had IR-1061, a commercially available water insoluble polymethine dye, embedded in a poly(acrylic acid) (PAA), an amphiphilic polymer, matrix. These nanoparticles also had a coating of the surfactant DSPE-mPEG (polyethylene glycol-conjugated phospholipid, ca. 5 kDa) for improved biocompatibility and longer circulation lifetimes. Using these IR-1061 nanoparticles, the inner organs and blood vessels of mice were imaged and the results matched the images previously obtained with inorganic carbon nanotubes and quantum dots.164 Most of the recent literature published on nanoparticle systems not only are biocompatible, fully biodegradable, and exhibit high signals but are also designed to target particular cells or tumors and avoid healthy cells. Specific details of these systems and examples will be discussed in the next section. Passive targeting of nanoparticles has also been recently used with self assembled D-α-tocopheryl polyethylene glycol 1000 succinate conjugated to cis-aconitic anhydride modified doxorubicin, a common chemotherapy drug. After the self assembly of the nanoparticles, the fluorophore chlorin e6 was loaded into the nanoparticles via encapsulation. The nanoparticle system as a whole is nonfluorescent. However, cis-aconitic anhydride modified doxorubicin is acid sensitive, and upon cellular internalization and subsequent transport to the acidic lysosomes, the amide linkage is hydrolyzed and the fluorophore and drug are released. Successful photodynamic therapy was confirmed with this system.165 Self-assembly of nanoparticles was also utilized to yield tetraphenylethene fluorophore nanoparticles. Tetraphenylethene exhibits aggregation induced emission, making it an ideal candidate for fluorescence imaging. Further, the tetraphenylethene self-assembled nanoparticles exhibited very good singlet oxygen generation as well as emission in the NIR region. Tetraphenylethene nanoparticles showed a maximum absorption of 450 nm with a maximum emission of approximately 700 nm.166 Another study investigated PEGylated graphene oxide nanoparticles with encapsulated sinoporphyrin sodium (DVDMS). These nanoparticles exhibited increased accumulation in cancer cells than DVDMS alone. Further, the nanoparticles showed more efficient apoptosis/necrosis via photodynamic therapy when compared to that induced by DVDMS alone.167 Nanoparticles made from derivative of anthraquinone, a fluorophore exhibiting aggregation induced emission, via a reprecipitation method were synthesized. These particles showed low cytotoxicity and good stability in cells (A549 cell line) for 15 days over six generations. This system is a good candidate for performing fluorescence imaging of cancer over an extended period of time.168
 | ||
| Fig. 8 Real-time fluorescence imaging of BF2 chelated tetraarylazadipyrromethenes (near infrared dye) conjugated poly(styrene-co-methacrylic acid) particles uptake into HEK293T cells (scale bar 10 μm) (reprinted with permission from ref. 163 Copyright 2011 American Chemical Society). | ||
4 Targeting
Up to this point examples of passive targeting of tumors in fluorescence imaging have only been discussed. Encapsulating or conjugating small molecule fluorophores to nanoparticles allows the particles to preferentially accumulate in tumors due to the EPR effect (cf. Fig. 9).100,101 In this case, the size is used to passively target tumors. Apart from size, modifying the surface charge of the nanoparticles is an additional way to passively target the tumors. The surface charge of tumors is highly negative when compared to normal cells,169 so if the surface of the nanoparticles is modified to be more cationic, then the nanoparticles bind electrostatically to the negatively charged phospholipid head groups expressed on tumors and in turn preferentially accumulate in tumors.170–173 However, passive targeting is limited; therefore, it is necessary to add an active targeting component to the nanoparticles so that they affect tumors and avoid healthy cells (cf. Fig. 9). An ideal target should be universally and uniquely expressed by tumors. In most cases the target has an overexpression of a particular cell surface marker in tumors.170 The overexpression of the target group facilitates increased binding of the targeting agent, which increases the cellular uptake of the targeting agent.106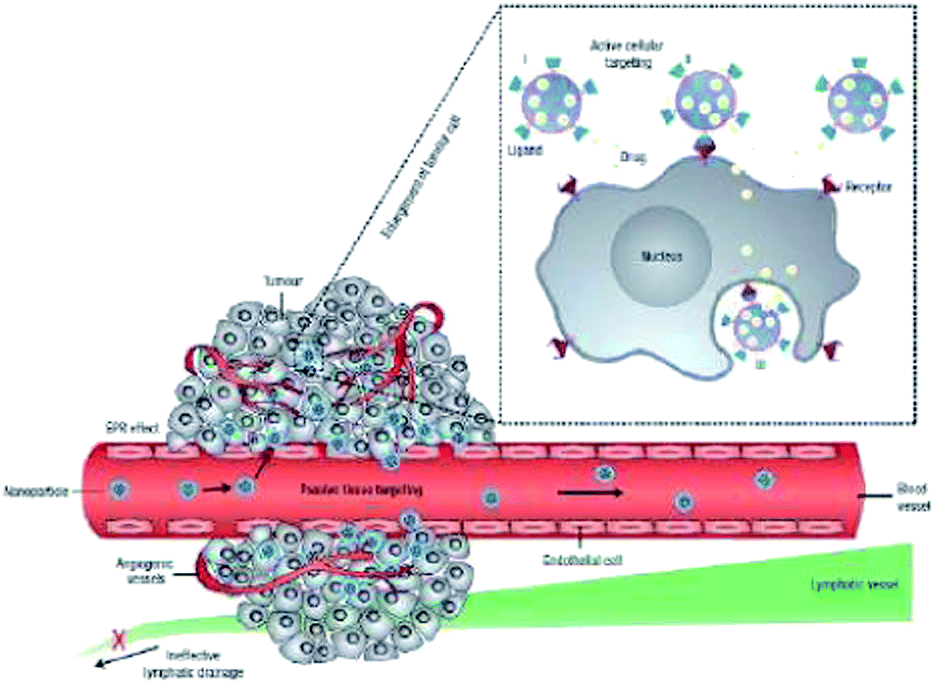 | ||
| Fig. 9 Cartoon depicting different mechanisms of drug delivery to cancer cells with nanocarriers. Polymeric nanoparticles are shown as representative nanocarriers (circles). Passive targeting is achieved by extravasation of nanoparticles through the enhanced permeation and retention (EPR) effect. Active cellular targeting (inset) can be achieved by tagging the nanoparticle surface with targeting ligands that specifically bind with a receptor on the cell surface. The nanoparticles can either (i) release their cargo when in proximity to the target cells; (ii) attach to the cell membrane and function as an extracellular sustained-release drug reservoir; or (iii) internalize into the cell (reprinted with permission from ref. 100 Copyright 2007 Nature Publishing Group). | ||
In ligand-based targeting, the ligand binds specifically to a receptor that is overexpressed by tumor cells. The ligands used vary from small molecules, vitamins, carbohydrates, peptides, antibodies, proteins, and nucleic acids.100,170,174,175 A system was developed to detect nitroreductase (NTR), which is overexpressed in tumors experiencing hypoxia. Hypoxia occurs when tumors are deoxygenitated; in the tumor, the oxygen level is 0–4%, which inhibits many therapies that require oxygen in order to be effective. The system for the detection of NTR consists of para-nitro benzoate functionalized Cy7 cyanine dye. The nitro-benzoate linker performs the detection of NTR, while the cyanine dye provides the fluorescence signal. The aromatic nitrogen group binds to NTR through strong hydrogen bonds. Once the probe is bound to the NTR, the system, which was originally nonfluorescent, becomes fluorescent due to the changing strength of the electron withdrawing group upon binding, serving as an optical indicator of NTR.176 Another recent development in ligand-based targeting is the use of a GE-137 peptide that binds to human tyrosine kinase c-Met; human tyrosine kinase is prominent in pre-cancerous colorectal polyps. In this targeting system, GE-137 is conjugated to a cyanine dye, which fluoresces allowing for an optical cue for physicians performing fluorescence colonoscopies that there is a pre-cancerous polyp in the colon. Fluorescence colonoscopies led to 18% more polyps being detected by physicians than when only white light colonoscopies were performed on the same patients.177 Another study used a phthalocyanine dye that had been modified via streptavidin–biotin chemistry to have an αvβ6 integrin targeting ligand. It was found through in vitro, in vivo, and ex vivo fluorescence and optical imaging and ex vivo PET scans that the targeted phthalocyanine system did successfully bind to αvβ6 integrin, which is widely upregulated in cancers, especially pancreatic cancer.178 In another investigation, silica nanoparticles with encapsulated salicylaldehyde were surface modified with a DNA aptamer that selectively binds to nucleolin. MCF-7 cancer cells, which overexpress nucleolin, in comparison with normal MCF-10A cells were used to evaluate the targeting efficiency of the functionalized nanoparticles. In vitro studies showed that the nanoparticles preferentially accumulated in the cancer cells and effective fluorescence imaging was achieved with the nanoparticles via aggregation induced emission.179
Commonly targeted surface cell receptors are transferrin, folate, epidermal growth factor receptors (EGFRs), and glycoproteins.170,180–185 A study was performed where a targeting ligand was conjugated to a sulfur substituted BODIPY derivative. The target was γ-glutamyltranspeptidase (GGT), which is overexpressed on the plasma membrane of tumor cells and serves to cleave glutamate. The probe, when in the presence of GGT, causes the enzymically triggered conversion of the sulfur substituted BODIPY derivative to the amino-substituted BODIPY, which causes a large shift in the maximum emission of the BODIPY. In this way, GGT levels can be monitored and tumors can be imaged.186 Early 2016 saw the development of a NIR-II targeted PEGylated fluorophore (CH1055) probe that was 90% cleared by the kidneys within 24 h. The probe was surface functionalized with an anti-EGFR Affibody. The fluorophore exhibits a desirable large Stokes shift of 175 nm with a maximum absorption of 750 nm and a maximum emission of 1055 nm. In immunodeficient nude mice, successful imaging, targeting, and cancer ablation was achieved.187 Silica nanoparticles were used to encapsulate palladium(II) tetraphenyltetrabenzoporphyrin (donor) and perylene (acceptor) or to encapsulate palladium(II) tetraphenyltetrabenzoporphyrin (donor) and 9,10-bis-(phenylethynyl)anthracene (BPEA) (acceptor) to yield two distinct triplet–triplet annihilation upconversion fluorophore pairs, which emit either green or blue. Further, the nanoparticle surface was covalently modified with various peptides or antibodies for active targeting. It should be noted that upconversion is an anti-Stokes phenomenon in which at least 2 photons exhibiting low frequency are combined and converted into one photon with a high frequency.188
In tumor endothelium targeting, the endothelial cells are targeted to prevent angiogenesis (production of new blood vessels), as it plays an important role in regulating cancer growth. Targeting vascular endothelial growth factors (VEGF) receptor, αvβ3 integrin, vascular cell adhesion molecule-1 (VCAM-1), and matrix metalloproteinases (MMP) are some examples of tumor endothelium targeting.170,189–193 One study in tumor endothelium targeting employed poly(lactic acid) (PLA) nanoparticles that had encapsulated Endostar, a recombinant human endostain that is known to inhibit angiogenesis. Further, the PLA nanoparticles had been surface modified with a fluorophore, namely NIR IRdye 800CW, and a GX1 peptide, which has been shown to bind to the endothelium. It was found that the complex did bind to the endothelium through the in vivo fluorescence molecular imaging of mice with colorectal tumors. It should also be noted that functionalized PLA nanoparticles accumulated better and resulted in a higher fluorescence signal than free fluorophore, IRdye 800CW, alone.194 It was found that the BODIPY fluorophores synthesized by Kim and colleagues could be successfully encapsulated in silica (SiO2) nanoparticles. Then the surface of the silica nanoparticles could be modified to have an arginine-glycine-aspartic (RGD) peptide that would selectively target αvβ3 integrin, which lead to enhanced fluorescence imaging with higher signal to background ratios when compared to the free fluorophores alone.98
In the majority of the applications, the targeting agent is not used by itself but is attached to a fluorophore or a nanocarrier to improve the accumulation. Most of these targeting agents are attached via a simple covalent conjugation technique or through electrostatic interactions. Attaching the targeting agent directly to the small molecule fluorophore compromises the emission and other properties of the probe, so attaching the targeting agents to the actual nanocarriers, which does not alter the properties of the fluorophore, is much preferred.107 In addition, conjugating the targeting agent to the polymeric nanocarrier overcomes weak reproducibility and poor control of tuning the number of targeting agents attached to the particle.132 Once the polymeric particle is conjugated with the desired amount of the targeting agent, they can be utilized to accumulate in a specific tumor for imaging applications.
Zheng and colleagues developed a novel ICG containing phospholipid-polyethylene glycol (ICG-PL-PEG) based micelle. They attached two targeting agents, a small molecule, namely folic acid (FA), and a large protein called integrin αvβ3 monoclonal antibody (mAb), to the ICG-PL-PEG micelles and displayed their target specificity using three different cell lines. The three cell lines used were EMT6 (murine mammary tumor cells), U87-MG (human glioblastoma cancer cells) and MCF-7 (human breast cancer cells). Laser scanning confocal microscopy, flow cytometry, and other in vitro experiments were employed to confirm that the targeting probe was mainly internalized into cells via ligand–receptor or antigen–antibody mediated endocytosis pathway. In addition, according to their data the amount of targeting agent conjugated to the ICG-PL-PEG micelle does not affect the integrity and properties of the nanoprobe.107
Pang and colleagues developed a 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) (fluorophore) loaded methoxy poly(ethylene glycol)-poly(σ-caprolactone) based polymersome. Apart from the fluorophore the polymersome was loaded with drugs, doxorubicin (Dox) and tetrandrine (Tet), to give a therapeutic functionality to the system. These multifunctional polymersomes were conjugated with a targeting agent called Lactoferrin (Lf) and this polymersome was referred to as Lf-PO-Dox/Tet-DiR. Lf is a novel brain targeting ligand, which enables drug-loaded nanocarriers to transport across the blood–brain barrier (BBB) with higher efficiency when compared to transferrin, another targeting agent. In vitro studies in C6 cells (rat glial tumor cells) were utilized to show that Lf-PO-Dox/Tet-DiR polymersomes were uptaken through receptor-mediated endocytosis, while the free Dox accumulated mainly through a diffusion mechanism. Moreover, in vivo studies performed in glioma model rats further confirmed that the Lf-PO-DiR crossed the BBB and had an increased accumulated at the tumor site, which is indicated by the strong increase in fluorescence at the site of the tumor (cf. Fig. 10). Overall, they constructed a promising targeted nanoprobe for diagnosis and therapy of gliomas.124,195
 | ||
| Fig. 10 (a) Design of lactoferrin-conjugated biodegradable polymersome for glioma targeting. Polymersomes are loaded with doxorubicin (Dox), an antitumor drug, and tetrandrine (Tet), a multi drug resistance (MDR) inhibitor, at the same time. Lactoferrin (Lf) was conjugated on the surface of polymersomes for two reasons: (1) to act as a glioma targeting ligand and (2) to help overcome the obstruction of the blood–brain-barrier (BBB). (b) Accumulation of targeted polymersomes in glioma (EPR effect and overcoming BBB) was shown through fluorescence imaging. (c) The specific interaction of Lf-conjugated polymersomes with glioma cells was shown by an improvement in the survival rate of rats treated with the system. The improved survival rate is a direct result of the Lf-conjugated polymersomes improving the therapeutic effect from chemotherapy (reprinted with permission from ref. 124 Copyright 2012 Wiley Periodicals Inc.). | ||
Le Droumaguet and colleagues developed a rhodamine B (fluorophore) functionalized poly(alkyl cyanoacrylate) (RhB-PACA) nanoparticles for fluorescence imaging. The RhB-PACA nanoparticles were surface functionalized with biologically active targeting ligands, such as biotin, curcumin derivatives, and a variety of antibodies. In vitro studies were performed in two cancer cell lines, MCF7 (human breast adenocarcinoma cells) and M109 (murine lung cancer cells), which both overexpress biotin receptors on their surfaces (cf. Fig. 11). The results obtained confirm that the biotin conjugated RhB-PACA nanoparticles were uptaken by the cells through a specific receptor mediated endocytic pathway as the fluorescence seen was mainly around the vesicles surrounding the nuclei. Moreover, the fluorescence signal of covalently conjugated rhodamine B to PACA nanoparticles was sharp and strong in comparison to the typical diffuse signal obtained when hydrophobic dyes are encapsulated. The specificity of biotin targeting strategy was further supported by in vitro studies performed in L1210 cells that do not overexpress biotin. The biotin-RhB-PACA nanoparticles did not show the same strong fluorescence in L1210 cells as shown in MCF7 and M109 cells.196
 | ||
| Fig. 11 Internalization of rhodamine B-tagged NPs. Fluorescence microscopy images of MCF7 cells show the cellular uptake of nonfunctionalized (N0) and biotin-functionalized (N1) NPs (red) after 5 h of incubation. The nuclei were stained with DAPI (blue) and phalloidin-fluorescein isothiocyanate (Ph-FITC, green) which was used to label F-actin. The last column of images is an overlay of all the staining (reprinted with permission from ref. 196 Copyright 2012 American Chemical Society). | ||
5 Activation
One of the issues noticed with polymer-core nanoparticles was that the emission of the probe was often initially quenched when dispersed in aqueous solutions due to the aggregation of the fluorophore on the surface of the particles.59 This drawback actually turned into an advantage for nanoparticulate systems because the probes are in a “turned off” fluorescence state under normal conditions and a “turned on” fluorescence state only under specific diseased conditions, which results in an enhanced tumor to background ratio.197 To this end, identifying different activation techniques for nanoprobes is necessary to develop activatable NIR nanoprobes. To date, various general activatable small molecule probes have been developed to image tumors and in most of these systems, the emission is “turned on” upon binding to a specific protein, enzyme, or receptor.197 Pham and colleagues reported a probe that is activated upon binding to matrix metalloproteinase 7 (MMP), a protease overexpressed in tumors.198 Tung and colleagues developed a NIR probe that activates when interacted with cathepsin D, which is another protease overexpressed in tumors.199 Urano and colleagues developed pH-activatable probes based on BODIPY fluorophore that had a cancer-targeting monoclonal antibody conjugated to it.200 There are only a few reported activatable nanoparticle systems. In 2011, Rungta and colleagues, used PA particles modified with an azide terminated ICG to demonstrate the activation of fluorescence when mixed with bovine serum albumin (BSA) (cf. Fig. 12).59 It is well established in literature that albumins can “turn on” the emission of aggregated fluorophores. Albumins bind to the hydrophobically aggregated fluorophore via a combination of hydrophobic, hydrogen bonding, and electrostatic interactions, which deaggregates the fluorophores and activates the emission.201 Later in 2011, Palma and colleagues demonstrated that the emission of BF2 chelated tetraarylazadipyrromethenes (near infrared dye) conjugated poly(styrene-co-methacrylic acid) particles can be activated by sodium dodecyl sulfate (SDS), a surfactant. In addition, they demonstrated the activation in vitro in MDAMB-231 (breast cancer), HEK293T (kidney), and CAKI-1 (renal cancer) cell lines. In this latter system, the phospholipids in the cell membrane, specifically lecithin is responsible for the activation and the mechanism is similar to that of SDS.163 These two strategies of activation are viable but are not specific. There has been advancement in the development of nanoparticle systems which respond to various stimuli and environments to activate emission (cf. Fig. 5),106,113,202 but less advancement has been seen for particle-based systems which activate emission upon binding to specific proteins or receptors that are overexpressed in tumors. It is expected that future endeavors in the field of tumor nanotheranostics will be in the direction of developing “smart” activatable NIR nanoparticles. For example, the well known fluorescence quenching effects of gold nanoparticles was employed by the Mirkin group203 to develop “nanoflares” which are designed to provide an intracellular emission signal that directly correlates with the concentration of a specific nucleic acid or other molecular target. These particles are oligonucleotide-functionalized gold nanoparticles that are tagged (i.e. hybridized) to short, fluorophore-labeled probes. Without a target, the fluorophore is close to the surface of the gold particle and the fluorescence is quenched, while binding to the target releases the fluorophore, resulting in a signal. Similarly, activation was achieved for a BODIPY probe covalently conjugated to N-benzyl-4-hydroxyaniline. N-Benzyl-4-hydroxyaniline is sensitive to nitric oxide, which is responsible for regulating a wide variety of physiological processes; however, when the amount of nitric oxide becomes unbalanced, it can be a cause or indicator of cancer. Therefore, detecting irregular levels of nitric oxide is desirable. Originally, the BODIPY/N-benzyl-4-hydroxyaniline conjugate is nonfluorescent due to N-benzyl-4-hydroxyaniline quenching the fluorescence of BODIPY. However, upon interaction with nitric oxide, the N-benzyl-4-hydroxyaniline undergoes a nitrosation reaction, which effectively activates the fluorescence of BODIPY and serves as a visual indicator of nitric oxide. Further, the amount of nitric oxide can be detected by monitoring the emission seen from the BODIPY fluorophore.204 Yuan and colleagues created a novel nanoprobe with a derivatized tetraphenylethylene covalently attached to 2,4-dinitrobenzenesulfonyl. It should be noted that tetraphenylethylene is a commonly used fluorophore exhibiting aggregation induced emission. However, this absorption of the fluorophore is in the UV range of the electromagnetic spectrum, which decreases the effectiveness of the fluorophore in fluorescence imaging. Therefore, tetraphenylethylene was modified to have a dicyanovinyl group and methoxy group, which act as a donor/acceptor pair which redshifts the absorption of the fluorophore. Upon reaction with 2,4-dinitrobenzenesulfonyl, the fluorescence of tetraphenylethylene is quenched. However, upon cellular uptake and interaction with biothiols, the quencher is cleaved from the fluorophore, which activates the fluorescence of tetraphenylethylene, and a high signal to noise ratio was observed. Tetraphenylethylene is a singlet oxygen generator and with MDA-MB-231 cells, cell death was observed. The nanoprobe was further functionalized with a cyclic RGD probe, providing active targeting for cells overexpressing αvβ3 integrin.205 Nanoparticles made from curcumin (Cur), a chemotherapy drug, with encapsulated perylene and 5,10,15,20-tetra(4-pyridyl) porphyrin (H2TPyP) (a donor–acceptor pair) were synthesized. The intact system, upon initial cellular internalization, have no therapeutic effect, and they have a red emission due to FRET between the fluorophores, which enhances the effect of photodynamic therapy due to the increased fluorescence emission cause by FRET. After some time, Cur nanoparticles begin to dissociate, treating cancer via chemotherapy, while the fluorescence of the system shifts from red to green. Cur emits green, and, due to the disintegration of the Cur particles, FRET is no longer achievable between perylene and H2TPyP. However, any part of the intact Cur particles will continue to emit red; therefore, this system can be used to estimate real time drug dosage.206 | ||
| Fig. 12 (a) Increase in the emission intensity ratio of PA-azICG-azPEG1K particles dispersed in a phosphate buffered solution (PBS) and after the addition of 0.014 mM BSA; the graph depicts the time evolution of the intensity at 819 nm relative to the initial intensity. The inset presents emission of particles after 2 min (○), 37 min (●), and 1174 min (∇). Excitation was at 710 nm; particle density was 1.259 × 1012 cm−3. (b) Optical image of emission intensity of PA-azICG-azPEG1K particles in deionized water (far left), PBS (center), and 2 h after the addition of 0.014 mM BSA to the PBS solution (far right); images were taken with a Caliper Xenogen IVIS Lumina II XR Instrument with excitation at 745 nm and emission was observed with an ICG emission filter; particle density of 1.259 × 1012 cm−3 (reprinted with permission from ref. 59 Copyright 2011 WILEY-VCH Verlag GmbH & Co. KGaA). | ||
6 Perspectives and conclusions
Strides have been made in ensuring that fluorescence imaging is an enhanced way to detect cancer in its early stages without the drawbacks of traditional methods. Great care has been taken in improving the fluorophores used for imaging, such as coupling them with nanocarriers, which has ameliorated many of the downfalls of the currently FDA approved fluorophores, such as ICG and photofrin. Further advancements have been made in the delivery of these fluorophores to the intended cancerous or pre-cancerous site to provide physicians with as much information about the tumor as possible while limiting the side effects for the patient (i.e. radiation exposure). Little doubt remains that these fluorophores and their nanocarriers will eventually revolutionize the way cancer is diagnosed and treated.Acknowledgements
The authors thank the Gregg-Graniteville Foundation and the National Science Foundation (DMR-1507266) for financial support.References
- S. Trabulo, A. M. Cardoso, T. Santos-Ferreira, A. L. Cardoso, S. Simoes and M. C. P. de Lima, Mol. Pharm., 2011, 8, 1120–1131 CrossRef CAS PubMed.
- Y. D. Jin, C. X. Jia, S. W. Huang, M. O'Donnell and X. H. Gao, Nat. Commun., 2010, 1, 1–8 CrossRef PubMed.
- T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545–580 CrossRef CAS PubMed.
- M. J. Paulus, S. S. Gleason, M. E. Easterly and C. J. Foltz, Lab. Anim., 2001, 30, 36–45 CAS.
- V. V. Mody, M. I. Nounou and M. Bikram, Adv. Drug Delivery Rev., 2009, 61, 795–807 CrossRef CAS PubMed.
- M. E. Phelps, Neurochem. Res., 1991, 16, 929–940 CrossRef CAS PubMed.
- S. M. Janib, A. S. Moses and J. A. MacKay, Adv. Drug Delivery Rev., 2010, 62, 1052–1063 CrossRef CAS PubMed.
- A. J. Beer and M. Schwaiger, Cancer Metastasis Rev., 2008, 27, 631–644 CrossRef CAS PubMed.
- M. R. Zalutsky, D. A. Reardon, O. R. Pozzi, G. Vaidyanathan and D. D. Bigner, Nucl. Med. Biol., 2007, 34, 779–785 CrossRef CAS PubMed.
- P. Debbage and W. Jaschke, Histochem. Cell Biol., 2008, 130, 845–875 CrossRef CAS PubMed.
- K. Park, S. Lee, E. Kang, K. Kim, K. Choi and I. C. Kwon, Adv. Funct. Mater., 2009, 19, 1553–1566 CrossRef CAS.
- D. J. Spergel, U. Kruth, D. R. Shimshek, R. Sprengel and P. H. Seeburg, Prog. Neurobiol., 2001, 63, 673–686 CrossRef CAS PubMed.
- R. Weissleder, C. H. Tung, U. Mahmood and A. Bogdanov, Nat. Biotechnol., 1999, 17, 375–378 CrossRef CAS PubMed.
- B. Chance, M. Cope, E. Gratton, N. Ramanujam and B. Tromberg, Rev. Sci. Instrum., 1998, 69, 3457–3481 CrossRef CAS.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 CrossRef CAS PubMed.
- N. Nasongkla, E. Bey, J. M. Ren, H. Ai, C. Khemtong, J. S. Guthi, S. F. Chin, A. D. Sherry, D. A. Boothman and J. M. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS PubMed.
- E. Lallana, F. Fernandez-Trillo, A. Sousa-Herves, R. Riguera and E. Fernandez-Megia, Pharm. Res., 2012, 29, 902–921 CrossRef CAS PubMed.
- H. P. Yap, A. P. R. Johnston, G. K. Such, Y. Yan and F. Caruso, Adv. Mater., 2009, 21, 4348–4352 CrossRef CAS PubMed.
- Z. H. Sheng, D. H. Hu, M. M. Xue, M. He, P. Gong and L. T. Cai, Nano-Micro Lett., 2013, 5, 145–150 CrossRef.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed.
- S. K. Sahoo and V. Labhasetwar, Drug Discovery Today, 2003, 8, 1112–1120 CrossRef CAS PubMed.
- B. Le Droumaguet, J. Nicolas, D. Brambilla, S. Mura, A. Maksimenko, L. De Kimpe, E. Salvati, C. Zona, C. Airoldi, M. Canovi, M. Gobbi, M. Noiray, B. La Ferla, F. Nicotra, W. Scheper, O. Flores, M. Masserini, K. Andrieux and P. Couvreur, ACS Nano, 2012, 6, 5866–5879 CrossRef CAS PubMed.
- J. K. Pokorski, K. Breitenkamp, L. O. Liepold, S. Qazi and M. G. Finn, J. Am. Chem. Soc., 2011, 133, 9242–9245 CrossRef CAS PubMed.
- J. W. Cui, Y. Yan, Y. J. Wang and F. Caruso, Adv. Funct. Mater., 2012, 22, 4718–4723 CrossRef CAS.
- J. Merian, J. Gravier, F. Navarro and I. Texier, Molecules, 2012, 17, 5564–5591 CrossRef CAS PubMed.
- H. Wada, H. Hyun, C. Vargas, J. Gravier, G. Park, S. Gioux, J. V. Frangioni, M. Henary and H. S. Choi, Theranostics, 2015, 5, 1–11 CrossRef CAS PubMed.
- S. L. Gibbs, Quant. Imag. Med. Surg., 2012, 2, 177–187 Search PubMed.
- Y. Ashitate, H. Hyun, S. H. Kim, J. H. Lee, M. Henary, J. V. Frangioni and H. S. Choi, Theranostics, 2014, 4, 693–700 CrossRef PubMed.
- S. L. Gibbs-Strauss, K. A. Nasr, K. M. Fish, O. Khullar, Y. Ashitate, T. M. Siclovan, B. F. Johnson, N. E. Barnhardt, C. A. T. Hehir and J. V. Frangioni, Mol. Imaging, 2011, 10, 91–101 CAS.
- H. S. Choi, S. L. Gibbs, J. H. Lee, S. H. Kim, Y. Ashitate, F. Liu, H. Hyun, G. Park, Y. Xie, S. Bae, M. Henary and J. V. Frangioni, Nat. Biotechnol., 2013, 31, 148–153 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- R. Weissleder and U. Mahmood, Radiology, 2001, 219, 316–333 CrossRef CAS PubMed.
- R. Alford, M. Ogawa, P. L. Choyke and H. Kobayashi, Mol. BioSyst., 2009, 5, 1279–1291 RSC.
- L. Peters, Ann. N. Y. Acad. Sci., 1948, 50, 117 CrossRef CAS PubMed.
- A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817–840 CrossRef CAS.
- J. J. Vos, J. K. G. Wietasch, A. R. Absalom, H. G. D. Hendriks and T. W. L. Scheeren, Anaesthesia, 2014, 69, 1364–1376 CrossRef CAS PubMed.
- J.-J. Lee, C.-F. Chang, J.-R. Sheu and T. Jayakumar, Curr. Pharm. Biotechnol., 2014, 15, 700–711 CAS.
- S. Mimura, Y. Ito, T. Nagayo, M. Ichii, H. Kato, H. Sakai, K. Goto, Y. Noguchi, H. Tanimura, Y. Nagai, S. Suzuki, Y. Hiki and Y. Hayata, Lasers Surg. Med., 1996, 19, 168–172 CrossRef CAS PubMed.
- A. K. H. Kwok, T. Y. Y. Lai, W. W. Y. Li, D. T. W. Yew and V. W. Y. Wong, Eye, 2004, 18, 882–888 CrossRef CAS PubMed.
- C. Chi, J. Ye, H. Ding, D. He, W. Huang, G.-J. Zhang and J. Tian, PLoS One, 2013, 8, 1–11 Search PubMed.
- G. Boniface and M. Azab, Eur. J. Canc. Care, 1999, 8, 25–30 Search PubMed.
- D. A. Bellnier, W. R. Greco, G. M. Loewen, H. Nava, A. R. Oseroff and T. J. Dougherty, Lasers Surg. Med., 2006, 38, 439–444 CrossRef PubMed.
- J. Merian, J. Gravier, F. Navarro and I. Texier, Molecules, 2012, 17, 5564–5591 CrossRef CAS PubMed.
- X. Yi, F. Wang, W. Qin, X. Yang and J. Yuan, Int. J. Nanomed., 2014, 9, 1347–1365 CrossRef PubMed.
- M. S. Murahari and M. C. Yergeri, Curr. Pharm. Des., 2013, 19, 4622–4640 CrossRef CAS PubMed.
- Q. T. Nguyen and R. Y. Tsien, Nat. Rev. Cancer, 2013, 13, 653–662 CrossRef CAS PubMed.
- A. B. Nepomnyashchii and A. J. Bard, Acc. Chem. Res., 2012, 45, 1844–1853 CrossRef CAS PubMed.
- G. Fan, L. Yang and Z. Chen, Front. Chem. Sci. Eng., 2014, 8, 405–417 CrossRef CAS.
- N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 6577–6595 CrossRef CAS.
- R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- S. L. Luo, E. L. Zhang, Y. P. Su, T. M. Cheng and C. M. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed.
- C. H. Quek and K. W. Leong, Nanomaterials, 2012, 2, 92–112 CrossRef CAS.
- J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef CAS PubMed.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2011 CrossRef CAS PubMed.
- N. Tyutyulkov, F. Dietz, A. Ivanova and K. Mullen, Dyes Pigm., 1999, 42, 215–222 CrossRef CAS.
- P. Rungta, Y. P. Bandera, R. D. Roeder, Y. C. Li, W. S. Baldwin, D. Sharma, M. G. Sehorn, I. Luzinov and S. H. Foulger, Macromol. Biosci., 2011, 11, 927–937 CrossRef CAS PubMed.
- T. Gorecki, G. Patonay, L. Strekowski, R. Chin and N. Salazar, J. Heterocycl. Chem., 1996, 33, 1871–1876 CrossRef CAS.
- G. Patonay, M. D. Antoine, S. Devanathan and L. Strekowski, Appl. Spectrosc., 1991, 45, 457–461 CrossRef CAS.
- R. R. Avirah, D. T. Jayaram, N. Adarsh and D. Ramaiah, Org. Biomol. Chem., 2012, 10, 911–920 CAS.
- A. Treibs and K. Jacob, Angew. Chem., Int. Ed., 1965, 4, 694–695 CrossRef.
- L. Hu, Z. Q. Yan and H. Y. Xu, RSC Adv., 2013, 3, 7667–7676 RSC.
- F.-P. Gao, Y.-X. Lin, L.-L. Li, Y. Liu, U. Mayerhoeffer, P. Spenst, J.-G. Su, J.-Y. Li, F. Wuerthner and H. Wang, Biomaterials, 2014, 35, 1004–1014 CrossRef CAS PubMed.
- E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289 CrossRef CAS PubMed.
- J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329–6338 RSC.
- K. Umezawa, D. Cittierio and K. Suzuki, Anal. Sci., 2008, 24, 213–217 CrossRef CAS PubMed.
- N. Barbero, C. Magistris, J. Park, D. Saccone, P. Quagliotto, R. Buscaino, C. Medana, C. Barolo and G. Viscardi, Org. Lett., 2015, 17, 3306–3309 CrossRef CAS PubMed.
- L. B. Josefsen and R. W. Boyle, Theranostics, 2012, 2, 916–966 CrossRef CAS PubMed.
- M. Hanack, M. Hees, P. Stihler, G. Winter and L. R. Subramanian, in Synthesis and Properties of Conducting Bridged Macrocyclic Metal Complexes, ed. T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Marcel Dekker Inc., New York, 1984, pp. 381–407 Search PubMed.
- S. Zhang, T. Abe, T. Iyoda and K. Nagai, Molecules, 2012, 17, 10801–10815 CrossRef CAS PubMed.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed.
- J. D. Huang, S. Q. Wang, P. C. Lo, W. P. Fong, W. H. Ko and D. K. P. Ng, New J. Chem., 2004, 28, 348–354 RSC.
- N. Sekkat, H. van den Bergh, T. Nyokong and N. Lange, Molecules, 2012, 17, 98–144 CrossRef CAS PubMed.
- J. F. Lovell and P. C. Lo, Theranostics, 2012, 2, 815–816 CrossRef PubMed.
- M. Salome Rodriguez-Morgade, M. E. Plonska-Brzezinska, A. J. Athans, E. Carbonell, G. de Miguel, D. M. Guldi, L. Echegoyen and T. Torres, J. Am. Chem. Soc., 2009, 131, 10484–10496 CrossRef PubMed.
- J.-p. Taquet, C. Frochot, V. Manneville and M. Barberi-Heyob, Curr. Med. Chem., 2007, 14, 1673–1687 CrossRef CAS PubMed.
- S. A. Gorman, S. B. Brown and J. Griffiths, J. Environ. Pathol., Toxicol. Oncol., 2006, 25, 79–108 CrossRef CAS.
- N. Sekkat, H. van den Bergh, T. Nyokong and N. Lange, Molecules, 2012, 17, 98–144 CrossRef CAS PubMed.
- Y. Rio, M. Salome Rodriguez-Morgade and T. Torres, Org. Biomol. Chem., 2008, 6, 1877–1894 CAS.
- A. Gunsel, A. T. Bilgicli, M. Kandaz, E. B. Orman and A. R. Ozkaya, Dyes Pigm., 2014, 102, 169–179 CrossRef CAS.
- K. Lang, J. Mosinger and D. M. Wagnerova, Coord. Chem. Rev., 2004, 248, 321–350 CrossRef CAS.
- J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef CAS PubMed.
- B. G. Ongarora, X. K. Hu, S. D. Verberne-Sutton, J. C. Garno and M. G. H. Vicente, Theranostics, 2012, 2, 850–870 CrossRef CAS PubMed.
- H. Dincer, H. Mert, B. N. Sen, A. Dag and S. Bayraktar, Dyes Pigm., 2013, 98, 246–254 CrossRef CAS.
- Y. Bandera, M. K. Burdette, J. A. Shetzline, R. Jenkins, S. E. Creager and S. H. Foulger, Dyes Pigm., 2016, 125, 72–79 CrossRef CAS.
- S. C. Karunakaran, P. S. S. Babu, B. Madhuri, B. Marydasan, A. K. Paul, A. S. Nair, K. S. Rao, A. Srinivasan, T. K. Chandrashekar, C. M. Rao, R. Pilai and D. Ramaiah, ACS Chem. Biol., 2013, 8, 127–132 CrossRef CAS PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- N. Yumita, Y. Iwase, K. Nishi, H. Komatsu, K. Takeda, K. Onodera, T. Fukai, T. Ikeda, S. Umemura, K. Okudaira and Y. Momose, Theranostics, 2012, 2, 880–888 CrossRef CAS PubMed.
- A. Treibs and F. H. Kreuzer, Justus Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS.
- Y. Ni and J. S. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 CAS.
- V. R. Donuru, S. L. Zhu, S. Green and H. Y. Liu, Polymer, 2010, 51, 5359–5368 CrossRef CAS.
- K. Umezawa, A. Matsui, Y. Nakamura, D. Citterio and K. Suzuki, Chem.–Eur. J., 2009, 15, 1096–1106 CrossRef CAS PubMed.
- K. Umezawa, Y. Nakamura, H. Makino, D. Citterio and K. Suzuki, J. Am. Chem. Soc., 2008, 130, 1550–1551 CrossRef CAS PubMed.
- D. Lakhe, K. K. Jairaj, M. Pradhan, U. Ladiwala and N. Agarwal, Tetrahedron Lett., 2014, 55, 7124–7129 CrossRef CAS.
- J. Bartelmess, M. Baldrighi, V. Nardone, E. Parisini, D. Buck, L. Echegoyen and S. Giordani, Chem.–Eur. J., 2015, 21, 9727–9732 CrossRef CAS PubMed.
- B. Kim, X. Yue, B. Sui, X. Zhang, Y. Xiao, M. V. Bondar, J. Sawada, M. Komatsu and K. D. Belfield, Eur. J. Org. Chem., 2015, 5563–5571 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- A. Fernandez-Fernandez, R. Manchanda and A. J. McGoron, Appl. Biochem. Biotechnol., 2011, 165, 1628–1651 CrossRef CAS PubMed.
- Z. L. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- G. Birrenbach and P. P. Speiser, J. Pharm. Sci., 1976, 65, 1763–1766 CrossRef CAS PubMed.
- L. W. Seymour, Y. Miyamoto, H. Maeda, M. Brereton, J. Strohalm, K. Ulbrich and R. Duncan, Eur. J. Cancer, 1995, 31A, 766–770 CrossRef CAS PubMed.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- X. H. Zheng, D. Xing, F. F. Zhou, B. Y. Wu and W. R. Chen, Mol. Pharm., 2011, 8, 447–456 CrossRef CAS PubMed.
- Y. Z. Chen, P. Z. Chen, H. Q. Peng, Y. Zhao, H. Y. Ding, L. Z. Wu, C. H. Tung and Q. Z. Yang, Chem. Commun., 2013, 49, 5877–5879 RSC.
- B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed.
- H. Tanisaka, S. Kizaka-Kondoh, A. Makino, S. Tanaka, M. Hiraoka and S. Kimura, Bioconjugate Chem., 2008, 19, 109–117 CrossRef CAS PubMed.
- V. B. Rodriguez, S. M. Henry, A. S. Hoffman, P. S. Stayton, X. D. Li and S. H. Pun, J. Biomed. Opt., 2008, 13, 1–10 CrossRef PubMed.
- S. Y. Zhang and Y. Zhao, Macromolecules, 2010, 43, 4020–4022 CrossRef CAS.
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel, M. Schmidt and A. Chilkoti, J. Am. Chem. Soc., 2008, 130, 687–694 CrossRef CAS PubMed.
- W. Kim, C. Brady and E. L. Chaikof, Acta Biomater., 2012, 8, 2476–2482 CrossRef CAS PubMed.
- V. Mundra, Y. Peng, S. Rana, A. Natarajan and R. I. Mahato, J. Controlled Release, 2015, 220, 130–140 CrossRef CAS PubMed.
- P. P. Ghoroghchian, P. R. Frail, K. Susumu, D. Blessington, A. K. Brannan, F. S. Bates, B. Chance, D. A. Hammer and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2922–2927 CrossRef CAS PubMed.
- Z. H. Li, L. Y. Wu, P. R. Hu, S. H. Han, T. Zhang, H. L. Fan, W. Jin, Q. H. Jin and Y. Mu, Nanoscale, 2012, 4, 7097–7105 RSC.
- P. P. Ghoroghchian, M. J. Therien and D. A. Hammer, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 156–167 CrossRef CAS PubMed.
- D. H. Levine, P. P. Ghoroghchian, J. Freudenberg, G. Zhang, M. J. Therien, M. I. Greene, D. A. Hammer and R. Murali, Methods, 2008, 46, 25–32 CrossRef CAS PubMed.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- M. Lomora, M. Garni, F. Itel, P. Tanner, M. Spulber and C. G. Palivan, Biomaterials, 2015, 53, 406–414 CrossRef CAS PubMed.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS PubMed.
- H. De Oliveira, J. Thevenot and S. Lecommandoux, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 525–546 CrossRef CAS PubMed.
- J. Z. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- M. Massignani, I. Canton, T. Sun, V. Hearnden, S. MacNeil, A. Blanazs, S. P. Armes, A. Lewis and G. Battaglia, PLoS One, 2010, 5, 1–11 Search PubMed.
- L. Quan, S. Liu, T. T. Sun, X. G. Guan, W. H. Lin, Z. G. Xie, Y. B. Huang, Y. Q. Wang and X. B. Jing, ACS Appl. Mater. Interfaces, 2014, 6, 16166–16173 CAS.
- G. Kania, U. Kwolek, K. Nakai, S.-I. Yusa, J. Bednar, T. Wojcik, S. Chlopicki, T. Skorka, M. Szuwarzynski, K. Szczubialka, M. Kepczynski and M. Nowakowska, J. Mater. Chem. B, 2015, 3, 5523–5531 RSC.
- R. Bakalova, D. Lazarova, B. Nikolova, S. Atanasova, G. Zlateva, Z. Zhelev and I. Aoki, Biotechnol. Biotechnol. Equip., 2015, 29, 175–180 CrossRef CAS PubMed.
- C. K. Wong, A. J. Laos, A. H. Soeriyadi, J. Wiedenmann, P. M. G. Curmi, J. J. Gooding, C. P. Marquis, M. H. Stenzel and P. Thordarson, Angew. Chem., Int. Ed., 2015, 54, 5317–5322 CrossRef CAS PubMed.
- R. Jetty, Y. P. Bandera, M. A. Daniele, D. Hanor, H. I. Hung, V. Ramshesh, M. F. Duperreault, A. L. Nieminen, J. J. Lemasters and S. H. Foulger, J. Mater. Chem. B, 2013, 1, 4542–4554 RSC.
- A. Vollrath, S. Schubert and U. S. Schubert, J. Mater. Chem. B, 2013, 1, 1994–2007 RSC.
- A. Schadlich, H. Caysa, T. Mueller, F. Tenambergen, C. Rose, A. Gopferich, J. Kuntsche and K. Mader, ACS Nano, 2011, 5, 8710–8720 CrossRef CAS PubMed.
- G. Storm, S. O. Belliot, T. Daemen and D. D. Lasic, Adv. Drug Delivery Rev., 1995, 17, 31–48 CrossRef CAS.
- S. M. Moghimi, Adv. Drug Delivery Rev., 1995, 17, 103–115 CrossRef CAS.
- P. Couvreur and C. Vauthier, Pharm. Res., 2006, 23, 1417–1450 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS PubMed.
- S. Schubert, J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581–1588 RSC.
- R. Reul, N. Tsapis, H. Hillaireau, L. Sancey, S. Mura, M. Recher, J. Nicolas, J. L. Coll and E. Fattal, Polym. Chem., 2012, 3, 694–702 RSC.
- V. Saxena, M. Sadoqi and J. Shao, J. Photochem. Photobiol., B, 2004, 74, 29–38 CrossRef CAS PubMed.
- V. Saxena, M. Sadoqi and J. Shao, Int. J. Pharm., 2004, 278, 293–301 CrossRef CAS PubMed.
- V. Saxena, M. Sadoqi and J. Shao, Int. J. Pharm., 2006, 308, 200–204 CrossRef CAS PubMed.
- A. Schadlich, C. Rose, J. Kuntsche, H. Caysa, T. Mueller, A. Gopferich and K. Mader, Pharm. Res., 2011, 28, 1995–2007 CrossRef PubMed.
- R. Tong, V. J. Coyle, L. Tang, A. M. Barger, T. M. Fan and J. J. Cheng, Microsc. Res. Tech., 2010, 73, 901–909 CrossRef CAS PubMed.
- E. Lallana, A. Sousa-Herves, F. Fernandez-Trillo, R. Riguera and E. Fernandez-Megia, Pharm. Res., 2012, 29, 1–34 CrossRef CAS PubMed.
- Q. Yang, J. Liu and X. Jiang, Progr. Chem., 2010, 22, 2377–2387 CAS.
- F. Musumeci, S. Schenone, A. Desogus, E. Nieddu, D. Deodato and L. Botta, Curr. Med. Chem., 2015, 22, 2022–2050 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- G. K. Such, S. T. Gunawan, K. Liang and F. Caruso, Macromol. Rapid Commun., 2013, 34, 894–902 CrossRef CAS PubMed.
- T. L. Ross, M. Honer, P. Y. H. Lam, T. L. Mindt, V. Groehn, R. Schibli, P. A. Schubiger and S. M. Ametamey, Bioconjugate Chem., 2008, 19, 2462–2470 CrossRef CAS PubMed.
- R. M. Desai, S. T. Koshy, S. A. Hilderbrand, D. J. Mooney and N. S. Joshi, Biomaterials, 2015, 50, 30–37 CrossRef CAS PubMed.
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064–1070 CrossRef CAS PubMed.
- M. J. Joralemon, R. K. O'Reilly, C. J. Hawker and K. L. Wooley, J. Am. Chem. Soc., 2005, 127, 16892–16899 CrossRef CAS PubMed.
- A. Kumar, U. J. Erasquin, G. Qin, K. Li and C. Cai, Chem. Commun., 2010, 46, 5746–5748 RSC.
- J. A. Opsteen, R. P. Brinkhuis, R. L. M. Teeuwen, D. W. P. M. Loewik and J. C. M. van Hest, Chem. Commun., 2007, 3136–3138 RSC.
- D. E. Achatz, G. Mezo, P. Kele and O. S. Wolfbeis, ChemBioChem, 2009, 10, 2316–2320 CrossRef CAS PubMed.
- S. Santra, C. Kaittanis, J. Grimm and J. M. Perez, Small, 2009, 5, 1862–1868 CrossRef CAS PubMed.
- H. M. Li, F. O. Cheng, A. M. Duft and A. Adronov, J. Am. Chem. Soc., 2005, 127, 14518–14524 CrossRef CAS PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192–3193 CrossRef CAS PubMed.
- Y. Xing, J. Zhao, P. S. Conti and K. Chen, Theranostics, 2014, 4, 290–306 CrossRef PubMed.
- K. Nwe and M. W. Brechbiel, Cancer Biother.Radiopharm., 2009, 24, 289–302 CrossRef CAS PubMed.
- A. Palma, L. A. Alvarez, D. Scholz, D. O. Frimannsson, M. Grossi, S. J. Quinn and D. F. O'Shea, J. Am. Chem. Soc., 2011, 133, 19618–19621 CrossRef CAS PubMed.
- Z. M. Tao, G. S. Hong, C. Shinji, C. X. Chen, S. Diao, A. L. Antaris, B. Zhang, Y. P. Zou and H. J. Dai, Angew. Chem., Int. Ed., 2013, 52, 13002–13006 CrossRef CAS PubMed.
- W. Hou, X. Zhao, X. Qian, F. Pan, C. Zhang, Y. Yang, J. Martinez de la Fuente and D. Cui, Nanoscale, 2016, 8, 104–116 RSC.
- D. T. Jayaram, S. Ramos-Romero, B. H. Shankar, C. Garrido, N. Rubio, L. Sanchez-Cid, S. Borros Gomez, J. Blanco and D. Ramaiah, ACS Chem. Biol., 2016, 11, 104–112 CrossRef CAS PubMed.
- X. Yan, G. Niu, J. Lin, A. J. Jin, H. Hu, Y. Tang, Y. Zhang, A. Wu, J. Lu, S. Zhang, P. Huang, B. Shen and X. Chen, Biomaterials, 2015, 42, 94–102 CrossRef CAS PubMed.
- J. Zhang, R. Chen, Z. Zhu, C. Adachi, X. Zhang and C.-S. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 26266–26274 CAS.
- A. M. James, E. J. Ambrose and J. H. Lowick, Nature, 1956, 177, 576–577 CrossRef CAS PubMed.
- R. H. Prabhu, V. B. Patravale and M. D. Joshi, Int. J. Nanomed., 2015, 10, 1001–1018 CAS.
- S. Ran, A. Downes and P. E. Thorpe, Cancer Res., 2002, 62, 6132–6140 CAS.
- A. Asati, S. Santra, C. Kaittanis and J. M. Perez, ACS Nano, 2010, 4, 5321–5331 CrossRef CAS PubMed.
- C. W. Beh, W. Y. Seow, Y. Wang, Y. Zhang, Z. Y. Ong, P. L. R. Ee and Y. Y. Yang, Biomacromolecules, 2009, 10, 41–48 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Preat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- D. B. Kirpotin, D. C. Drummond, Y. Shao, M. R. Shalaby, K. L. Hong, U. B. Nielsen, J. D. Marks, C. C. Benz and J. W. Park, Cancer Res., 2006, 66, 6732–6740 CrossRef CAS PubMed.
- Y. Li, Y. Sun, J. Li, Q. Su, W. Yuan, Y. Dai, C. Han, Q. Wang, W. Feng and F. Li, J. Am. Chem. Soc., 2015, 137, 6407–6416 CrossRef CAS PubMed.
- J. Burggraaf, I. M. C. Kamerling, P. B. Gordon, L. Schrier, M. L. de Kam, A. J. Kales, R. Bendiksen, B. Indrevoll, R. M. Bjerke, S. A. Moestue, S. Yazdanfar, A. M. J. Langers, M. Swaerd-Nordmo, G. Torheim, M. V. Warren, H. Morreau, P. W. Voorneveld, T. Buckle, F. W. B. van Leeuwen, L.-I. Odegardstuen, G. T. Dalsgaard, A. Healey and J. C. H. Hardwick, Nat. Med., 2015, 21, 955–961 CrossRef CAS PubMed.
- D. Gao, L. Gao, C. Zhang, H. Liu, B. Jia, Z. Zhu, F. Wang and Z. Liu, Biomaterials, 2015, 53, 229–238 CrossRef CAS PubMed.
- X. Wang, P. Song, L. Peng, A. Tong and Y. Xiang, ACS Appl. Mater. Interfaces, 2016, 8, 609–616 CAS.
- T. R. Daniels, T. Delgado, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 159–176 CrossRef CAS PubMed.
- P. S. Low and S. A. Kularatne, Curr. Opin. Chem. Biol., 2009, 13, 256–262 CrossRef CAS PubMed.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef CAS PubMed.
- J. J. Laskin and A. B. Sandier, Cancer Treat. Rev., 2004, 30, 1–17 CrossRef CAS PubMed.
- M. Scaltriti and J. Baselga, Cancer Treat. Rev., 2006, 12, 5268–5272 CAS.
- T. Minko, Adv. Drug Delivery Rev., 2004, 56, 491–509 CrossRef CAS PubMed.
- F. Wang, Y. Zhu, L. Zhou, L. Pan, Z. Cui, Q. Fei, S. Luo, D. Pan, Q. Huang, R. Wang, C. Zhao, H. Tian and C. Fan, Angew. Chem., Int. Ed., 2015, 54, 7349–7353 CrossRef CAS PubMed.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2016, 15, 235–243 CrossRef CAS PubMed.
- O. S. Kwon, H. S. Song, J. Conde, H.-I. Kim, N. Artzi and J.-H. Kim, ACS Nano, 2016, 10, 1512–1521 CrossRef CAS PubMed.
- T. Lammers, W. E. Hennink and G. Storm, Br. J. Cancer, 2008, 99, 392–397 CrossRef CAS PubMed.
- P. Carmeliet, Oncology, 2005, 69, 4–10 CrossRef CAS PubMed.
- J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9–22 CrossRef CAS PubMed.
- L. Osborn, C. Hession, R. Tizard, C. Vassallo, S. Luhowskyj, G. Chirosso and R. Lobb, Cell, 1989, 59, 1203–1211 CrossRef CAS PubMed.
- R. Roy, J. Yang and M. A. Moses, J. Clin. Oncol., 2009, 27, 5287–5297 CrossRef CAS PubMed.
- Y. Du, Q. Zhang, L. Jing, X. Liang, C. Chi, Y. Li, X. Yang, Z. Dai and J. Tian, Int. J. Nanomed., 2015, 10, 3791–3802 CrossRef CAS PubMed.
- Z. Pang, L. Feng, R. Hua, J. Chen, H. Gao, S. Pan, X. Jiang and P. Zhang, Mol. Pharm., 2010, 7, 1995–2005 CrossRef CAS PubMed.
- B. Le Droumaguet, J. Nicolas, D. Brambilla, S. Mura, A. Maksimenko, L. De Kimpe, E. Salvati, C. Zona, C. Airoldi, M. Canovi, M. Gobbi, M. Noiray, B. La Ferla, F. Nicotra, W. Scheper, O. Flores, M. Masserini, K. Andrieux and P. Couvreur, ACS Nano, 2012, 6, 5866–5879 CrossRef CAS PubMed.
- A. Yuan, J. H. Wu, X. L. Tang, L. L. Zhao, F. Xu and Y. Q. Hu, J. Pharm. Sci., 2013, 102, 6–28 CrossRef CAS PubMed.
- W. Pham, Y. D. Choi, R. Weissleder and C. H. Tung, Bioconjugate Chem., 2004, 15, 1403–1407 CrossRef CAS PubMed.
- C. H. Tung, U. Mahmood, S. Bredow and R. Weissleder, Cancer Res., 2000, 60, 4953–4958 CAS.
- Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104–109 CrossRef CAS PubMed.
- V. S. Jisha, K. T. Arun, M. Hariharan and D. Ramaiah, J. Phys. Chem. B, 2010, 114, 5912–5919 CrossRef CAS PubMed.
- K. Kim, M. Lee, H. Park, J. H. Kim, S. Kim, H. Chung, K. Choi, I. S. Kim, B. L. Seong and I. C. Kwon, J. Am. Chem. Soc., 2006, 128, 3490–3491 CrossRef CAS PubMed.
- A. E. Prigodich, D. S. Seferos, M. D. Massich, D. A. Giljohann, B. C. Lane and C. A. Mirkin, ACS Nano, 2009, 3, 2147–2152 CrossRef CAS PubMed.
- J. Miao, Y. Huo, X. Lv, Z. Li, H. Cao, H. Shi, Y. Shi and W. Guo, Biomaterials, 2016, 78, 11–19 CrossRef CAS PubMed.
- Y. Yuan, S. Xu, C.-J. Zhang, R. Zhang and B. Liu, J. Mater. Chem. B, 2016, 4, 169–176 RSC.
- J. Zhang, Y.-C. Liang, X. Lin, X. Zhu, L. Yan, S. Li, X. Yang, G. Zhu, A. L. Rogach, P. K. N. Yu, P. Shi, L.-C. Tu, C.-C. Chang, X. Zhang, X. Chen, W. Zhang and C.-S. Lee, ACS Nano, 2015, 9, 9741–9756 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |