
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dipole-induced asymmetric conduction in tunneling junctions comprising self-assembled monolayers†
Andrii
Kovalchuk
a,
David A.
Egger
c,
Tarek
Abu-Husein
b,
Egbert
Zojer
d,
Andreas
Terfort
b and
Ryan C.
Chiechi
 *a
*a
aStratingh Institute for Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: r.c.chiechi@rug.nl
bInstitut für Anorganishe und Analytische Chemie, Universität Frankfurt, Max-von-Laue-Straβe 7, 60438 Frankfurt, Germany
cDepartment of Materials and Interfaces, Weizmann Institute of Science, Rehovoth 76100, Israel
dInstitute of Solid State Physics, NAWI Graz, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria
First published on 12th July 2016
Abstract
This paper describes the observation of asymmetric conductance in the form of differing ratios of current density (J) as a function of voltage (|V|) in tunneling junctions comprising self-assembled monolayers on gold using eutectic Ga–In as a top contact. Monolayers comprising compounds with nearly identical physical and electronic properties show opposite directions of this asymmetry. We tested the statistical significance of the effect and ascribed it to the collective action of embedded dipoles arising from pyrimidyl groups that are arranged parallel or antiparallel to the transport direction. We ascribe the effect to the bias-induced (de)localization of the frontier states that mitigate transport.
Research efforts in molecular electronics fall into two broad and complementary experimental approaches to constructing tunneling junctions: single-molecule and large-area. The principal goal of the former is to develop a fundamental understanding of electron transport through junctions comprising single molecules, a construct that is relatively straightforward to model. The principal goal of the latter is functionality on the device-level, utilizing ensembles of molecules, usually in the form of self-assembled monolayers (SAMs), to define the properties and the smallest dimension of the junction.1 There are many phenomena that are unique to single-molecule junctions and that cannot be replicated in SAM-junctions; for example, contacting a molecule in different positions along its long axis,2 modulating conductance by changing rupture mechanics3,4 and utilizing anchoring groups that bind to electrodes through weak, non-covalent interactions.5 Likewise, SAMs exhibit collective effects that cannot be observed at the single-molecule level; for example, odd–even effects driven by the conformation of close-packed alkyl chains6–8 and the collective action of molecular dipoles affecting transition voltages (Vtrans) by shifting vacuum levels.9,10 Although single-molecule junctions are generally studied in greater detail, the technological relevance of SAM-junctions is more apparent and mature.11 Here, we report the asymmetric conduction in tunneling junctions using eutectic Ga–In (EGaIn) top-contacts12 that is driven by the collective action of dipole moments embedded in a SAM comprising molecules of identical empirical formula, frontier orbital energies (for isolated molecules) and interfaces with the electrodes (Fig. 1 on the left).
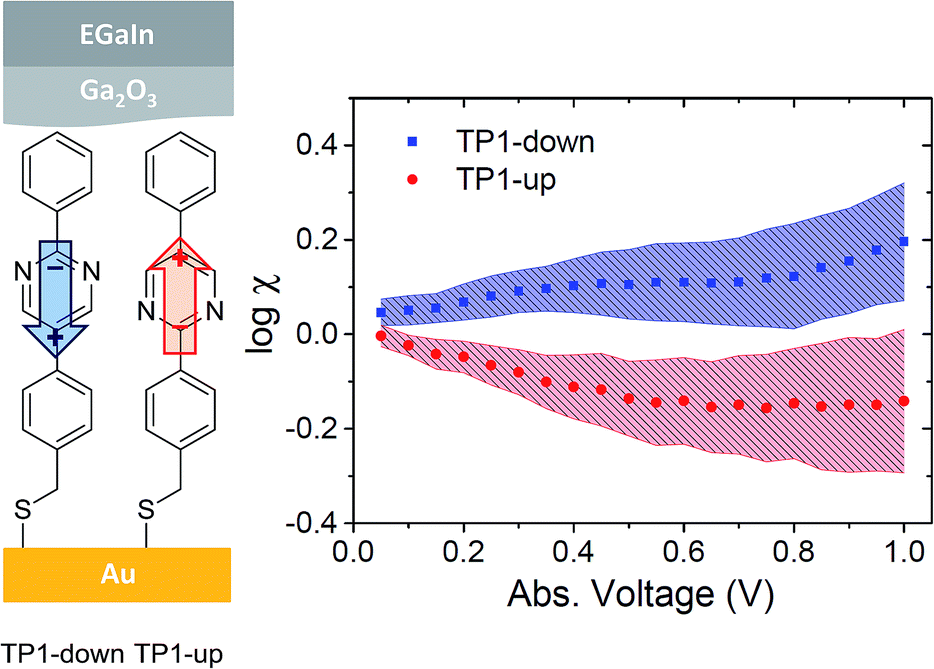 | ||
Fig. 1 Left: Schematic of a junction with two pyrimidyl-containing compounds (TP1-down and TP1-up) in junctions with Au and EGaIn electrodes. Arrows indicate directions of dipole moments associated with the embedded pyrimidine rings (from negative to positive). Right: Plots of asymmetry (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) χ, log of the ratio of |J| at each value of |V|) versus absolute voltage for SAMs of TP1-down (blue squares) and TP1-up (red circles) with 95% confidence intervals depicted as shaded areas. χ, log of the ratio of |J| at each value of |V|) versus absolute voltage for SAMs of TP1-down (blue squares) and TP1-up (red circles) with 95% confidence intervals depicted as shaded areas. | ||
Zhang et al. have shown theoretically that asymmetry in J/V characteristics can be caused by internal dipoles, if they induce an asymmetric voltage drop across the junction.13 We observed asymmetric J/V curves in junctions comprising SAMs of photosystem I (PSI) using both EGaIn and conducting-probe AFM.14 The direction of this asymmetry could not be ascribed to the electron-transport chain within the PSI complexes, rather it correlated to the net dipole induced by the peptide chains on the periphery. Although the mechanism seems obvious, it has received little experimental attention. And, while the results with PSI seem intuitive (and do induce a shift in vacuum level),15 the complexity of large protein complexes makes it difficult to isolate the underlying cause of the asymmetry. Thus, we turned to small molecules.
The SAMs we chose for this study (TP1-down, TP1-up; Fig. 1) have been extensively characterized by a number of techniques: high-resolution X-ray photoelectron spectroscopy (XPS), ellipsometry, infrared reflection absorption spectroscopy, near-edge X-ray absorption fine structure spectroscopy (NEXAFS), and scanning tunneling microscopy (STM).16 They exhibit nearly identical packing densities, thicknesses and tilt angles (see Table 1). The key difference is the orientation of a central pyrimidyl moiety that reverses the direction of its contribution to the net dipole moment along the direction of transport. These dipole moments act collectively in SAMs to affect the electrostatic profile across the junction17,18 (i.e., spanning the electrodes). We previously observed this effect experimentally in SAMs of three series of compounds as a shift in the work function (WF) of the bottom electrode16 and characterized its impact on tunneling charge-transport.9,10 Thus, the compounds used to form the SAMs in this study are regioisomers with nearly identical frontier orbitals (Fig. S7†) that form tunneling junctions of nearly identical geometries (Table 1).
| SAM | Effective thickness (nm) | Packing density (molecules per cm2) | Tilt angle |
|---|---|---|---|
| TP1-up | 1.73 ± 0.06 | 4.2 × 1014 | 18 ± 3° |
| TP1-down | 1.80 ± 0.07 | 4.3 × 1014 | 17 ± 3° |
We measured the J/V characteristics of SAMs of TP1-up and TP1-down on template-stripped gold (AuTS) by contacting them with sharp tips of EGaIn as described elsewhere.10 The absolute value of current density J is dominated by tunneling distance d (which is identical for both SAMs) and we observe statistically indistinguishable conductance at negative bias (Fig. S4†) and ordinary, bowl-shaped conductance associated with non-resonant tunneling (Fig. S5†). However, the magnitude of J differs at positive bias, creating asymmetry; i.e., TP1-down is more conductive at positive bias than negative bias and vice versa. To quantify this behavior we use an asymmetry parameter χ, defined as χ = |J+(V)/J−(V)|, and plot logχ versus |V| (Fig. 1). We show confidence intervals as error bars to highlight that values of logχ are statistically different across the full bias window; however, we cannot resolve whether Δlogχ saturates or continues to increase with bias. More detailed statistics are shown in the ESI.†
The absolute value of logχ is small compared to previous reports of SAM-based rectifiers based on localized π systems combined with n-alkyl chains.19,20 However, the mechanism of rectification in those SAMs requires an intrinsic molecular property—isolated, accessible states in the gap—and is therefore unrelated to our observation that the direction of asymmetry is correlated to the direction of the embedded dipole moment.
To explain the asymmetric J/V behavior we first compared it to the asymmetry in SAMs of PSI.14 According to that model, co-direction of the internal, perpendicular dipole moment of the SAM with the flow of electrons (which is incorrectly labeled as J in ref. 14) should result in higher current than anti-direction. Thus, TP1-up should be more conductive under positive bias and TP1-down at negative bias. However, we observe the opposite. To obtain further insight into the mechanism responsible for rectification, we calculated the transmission probability for TP1-up and TP1-down employing the model of single-molecule junctions consisting of an isolated TP1-up, respectively, TP1-down molecule and two Au clusters as electrodes. These simulations are not meant as models for AuTS/SAM//EGaIn junctions, rather they are meant to isolate the effects of the intrinsic electronic properties of TP1-up and TP1-down on transmission by examining a single molecule between ideal Au electrodes. If these transmission spectra differ, we cannot ascribe asymmetric conduction only to collective effects arising from packing into a SAM. The resulting transmission spectra (Fig. S6†) are essentially identical for the two molecules. Consistent with the discussions in ref. 10, 21 and 22, this result implies that the peculiar transport properties of TP1-up and TP1-down layers must arise solely from collective electrostatic effects induced by the parallel alignment of the pyrimidine dipoles within the SAMs.21,22 One can imagine that these effects induce asymmetric conduction either by affecting the level alignment in a peculiar way or by changing the nature of the molecular states such that, e.g., their spatial localization and the resulting transmission depend on the bias direction.
In order to clarify the effects of intermolecular effects in a SAM on transport, we first consider the details of the level-alignment at the interface(s). Explicitly modelling the behavior of the entire Au/SAM//Ga2O3/EGaIn is beyond the scope of this work, as the atomistic structure of the interface between Ga2O3 and the SAM is far from fully understood, particularly with respect to the strength of the coupling between the EGaIn electrode and the SAM. One can, however, draw insightful conclusions from considering two limiting cases; (i) a much weaker coupling between the SAM and EGaIn than between the SAM and Au and (ii) comparable coupling between the SAM and both electrodes. The former case is reminiscent of I/V measurements by STM, where the top electrode is decoupled from the SAM by vacuum and the bias direction determines whether transport occurs through occupied or unoccupied states. Non-zero values of logχ are observed when the Fermi level Ef of the Au electrode (which is strongly coupled to the SAM) does not lie in the exact center of the gap between these states. Since the values of logχ are positive for TP1-down and negative for TP1-up, this mechanism would require that the highest occupied π-state (HOPS) of SAMs of TP1-down lie closer to Ef than the lowest unoccupied π-state (LUPS) and vice versa for TP1-up. However, this situation is at odds with simulations of the projected density of states (PDOS) of these SAMs bonded to Au, where the occupied states are closer to Ef for both SAMs.10 The same study shows that the experimental values of |Vtrans| are comparable under positive and negative bias for both SAMs as well, which is only possible if transport takes place only through the HOPS. The other limiting case—assuming similar couplings between the SAM and the electrodes on both sides of the junction—is not unreasonable considering that the layer of Ga2O3 is on the order of 7 Å (ref. 23) and that there is a methylene spacer between the Au–thiolate anchor and the π-backbone. The aforementioned similarity in |Vtrans| also supports this hypothesis. In this scenario transport takes place through only one set of frontier states and, therefore, differences between the offsets of the HOPS and LUPS with the electrodes do not affect transport as they do in the case of weak coupling. Asymmetry is not expected in this case since the HOPS spans the junction and is assumed to couple similarly to both electrodes. Thus, transport through the LUPS is almost certainly not responsible for the observed trend in χ. In either case, there is no apparent reason why the level alignment should lead to non-zero values of logχ.
Having reasonably excluded level-alignment as the cause of asymmetry, we propose an alternative explanation based on bias-dependent de/localization of the frontier π states in SAMs of TP1-up and TP1-down. This electrostatic effect is related to the collective action of the dipoles of the pyrimidine rings spatially shifting the HOPS in TP1-down (TP1-up) towards (away from) the Au electrode. It is particularly evident when comparing the plane-averaged charge-density associated with the first peak in the PDOS of the SAM averaged over planes parallel to the surface, which is shown in Fig. 2 and was calculated with density functional theory using the VASP code,24 employing the HSE06 hybrid functional.25,26 That this de/localization is a consequence of collective electrostatics can be seen by comparing the plane-averaged charge-densities associated with the HOPS of isolated TP1-up and TP1-down molecules and the respective free-standing SAMs; the densities associated with the HOPS of isolated TP1-up and TP1-down molecules (orange lines, Fig. 2b and c) are virtually identical. The free-standing SAMs (grey shaded curves), however, differ from the isolated molecules and instead display essentially the same localization as the SAMs bound to Au (Fig. 2a). Thus, the spatial distribution of the orbitals through which transport occurs—the HOPS—is defined by the electrostatic environment of the SAM that results from the alignment of dipoles.
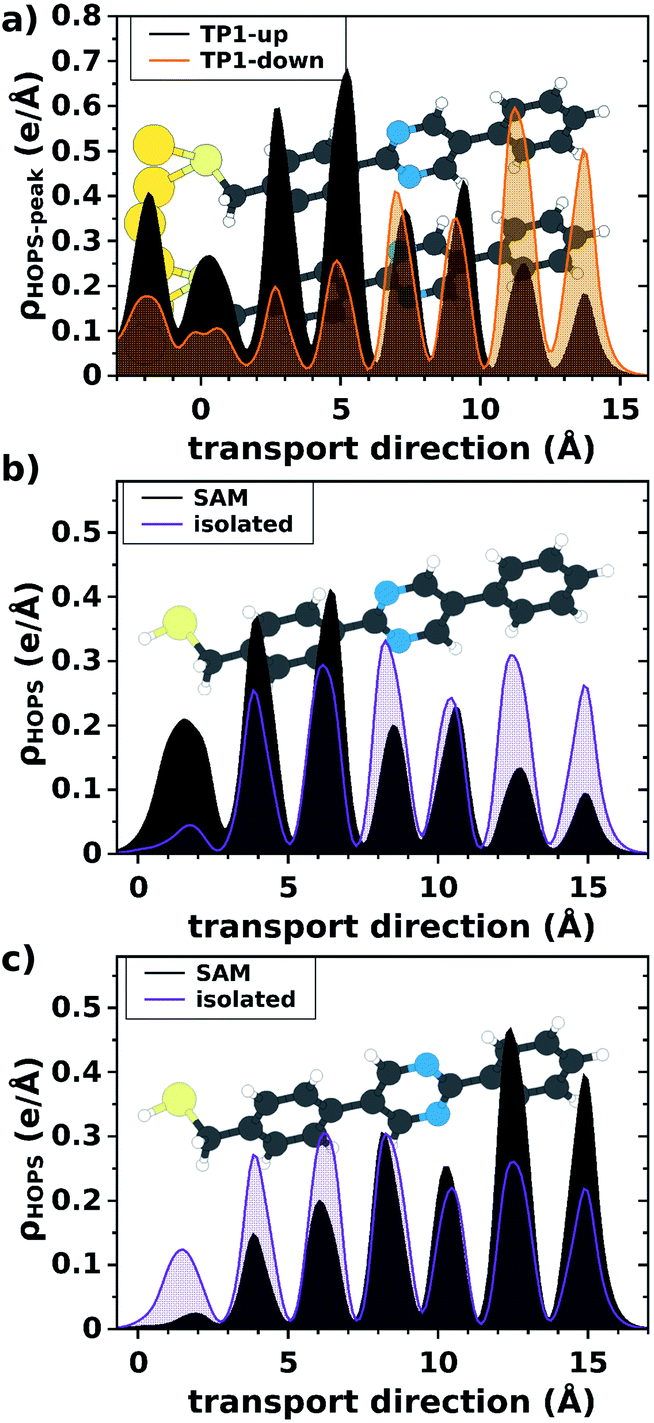 | ||
| Fig. 2 Plane-averaged charge density associated with the highest occupied peaks in the PDOS (derived from the molecular HOMO) of SAMs of TP1-up and TP1-down bonded to a Au electrode (panel a); the highest occupied π-state in the SAMs (without the electrode) and isolated molecules of TP1-up (panel b) and TP1-down (panel c). Note that for the SAMs without the Au electrode we plotted the charge density associated with the Γ-point of the corresponding band. The z-position of the first atom of the molecules/SAMs is chosen as the zero of the horizontal axis. Schematic representations of the structures27 are shown as a guide to the eye. All calculations on free-standing and Au-bonded SAMs rely on a periodic arrangement of the molecules. | ||
The distribution of the HOPS at zero bias alone does not explain the asymmetric conductance. There are two important considerations. First, the difference in couplings between the Au/SAM and SAM/EGaIn interfaces cannot be discounted entirely. Second, considering that the localization of the HOPS is a consequence of the dipole-induced potential steps within the SAMs, one should expect a further de/localization of the states depending on the direction of the applied bias; i.e., when the external field points in the same direction as the molecular dipole moment, localization should be reduced and vice versa. Assuming a direct correlation between delocalization and transmission, J should be higher in magnitude for TP1-down and lower for TP1-up at positive bias, which is exactly what we observe (Fig. 1).
To test the influence of bias on the direction-dependent localization of the states, we simulated the electronic structure of the two SAMs in an applied external field. This calculation can be done for the (hypothetical) free-standing SAMs in a relatively straightforward manner (see ESI† for details) and we know from the data shown in Fig. 2 that the presence of the Au-electrode does not qualitatively alter the localization. The choice of the free-standing SAM as a model system has the additional advantage that it allows SAM-related effects and interface-related effects to be separated and does not require guessing at the atomistic details of the SAM/Ga2O3 interface. The results of these simulations are shown in Fig. 3, confirming the hypothesis that the localization of states depends both on the direction of the dipole and the applied field (note that the definitions of the direction of the electric field, from + to −, and the dipole moment, from − to +, differ). Provided that the coupling at the side of the SAM where the charge-density is lower limits the current through the junction, positive bias (further localizing the HOPS) will lead to logχ < 0 in SAMs of TP1-up. Conversely, by delocalizing the HOPS in SAMs of TP1-down, it will lead to logχ > 0, in agreement with our experimental observations.
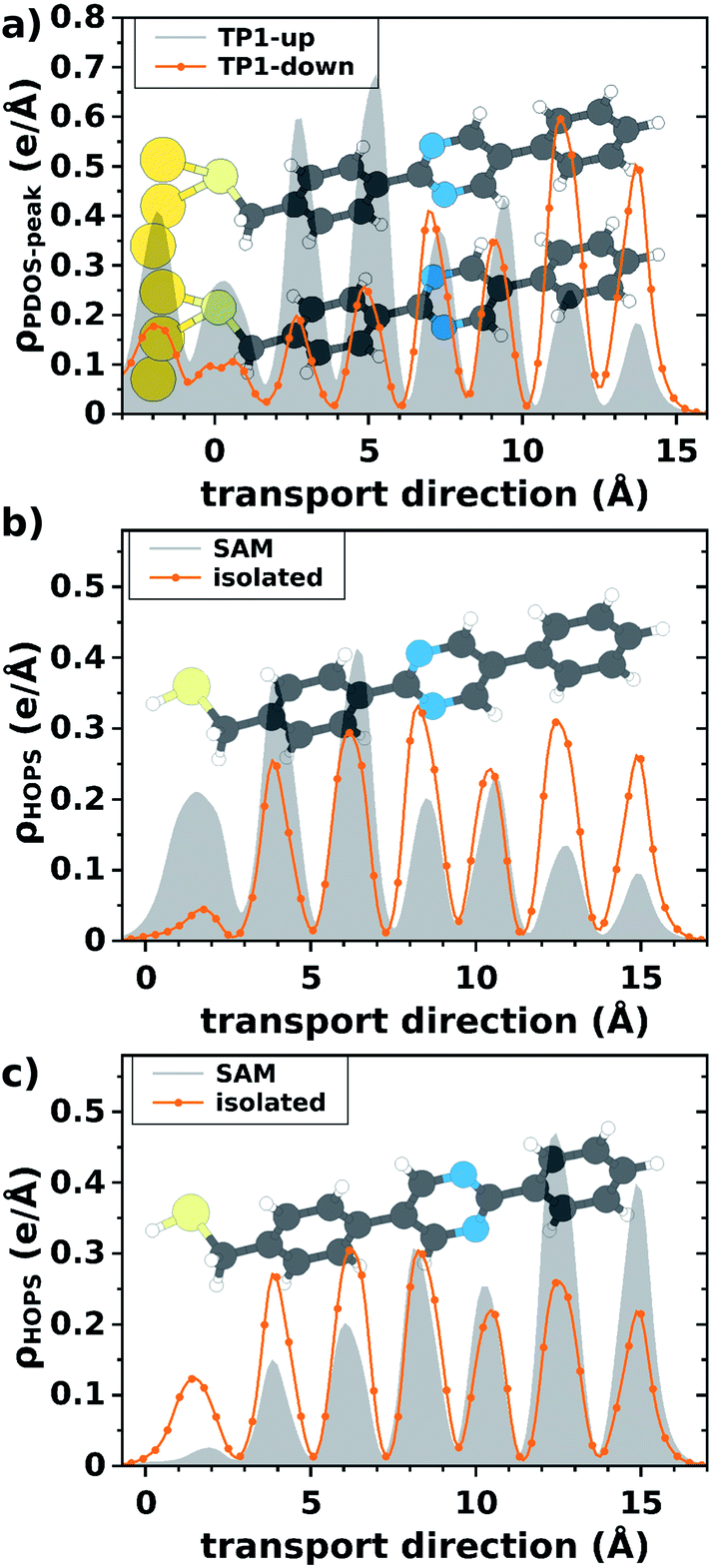 | ||
| Fig. 3 Plane-averaged charge density associated with the highest occupied π-state in the SAM (at the Γ-point; without Au electrode) for TP1-up (panel a) and TP1-down (panel b), as a function of an externally applied electric field. The z-coordinate of the first atom of the SAM is chosen as the zero for the horizontal axis. Schematic representations of the structures are shown as a guide to the eye. As screening effects are self-consistently considered in our simulations, we can estimate the average electric field within the SAM from the magnitude of the electrostatic energy in the vacuum regions above and below the free-standing monolayer. We find that screening is significant and that for an external field of 0.1 eV Å−1 the average internal field in the case of TP1-up amounts to 0.018 eV Å−1. Assuming an oxide thickness of 0.7 nm and a dielectric constant of 10 for β-Ga2O3,28 this field corresponds to a bias of 0.4 V—well within the experimental range. See ESI† for details. | ||
We observed a statistically significant asymmetry in tunneling charge-transport through SAMs of TP1-up and TP1-down. The direction of bias at which the conductance is higher correlates with the direction of the dipole moments arising from the central pyrimidine rings. The cause of this asymmetry is likely the result of the electrostatic profile of the SAMs arising from the collective effects of aligned dipoles affecting the de/localization of the frontier states. This mechanism is distinct from mechanisms that rely on isolated π systems or transport that occurs through both occupied and unoccupied states in that it is solely attributable to the supramolecular structure of the SAM.
Acknowledgements
AK and RCC acknowledge the European Research Council for the ERC Starting Grant 335473 (MOLECSYNCON). EZ acknowledges financial support from the Austrian Science Fund (FWF): I2081-N20. DAE acknowledges financial support from the Austrian Science Fund (FWF): J3608-N20. The computational results presented have been mostly achieved using the Vienna Scientific Cluster (VSC) and the clusters of the division for high-performance computing at the Graz University of Technology.References
- Y. Zhang, Z. Zhao, D. Fracasso and R. C. Chiechi, Isr. J. Chem., 2014, 54, 513–533 CrossRef CAS.
- D. Miguel, L. Á. de Cienfuegos, A. Martín-Lasanta, S. P. Morcillo, L. A. Zotti, E. Leary, M. Bürkle, Y. Asai, R. Jurado, D. J. Cárdenas, G. Rubio-Bollinger, N. Agraït, J. M. Cuerva and M. T. González, J. Am. Chem. Soc., 2015, 137, 13818–13826 CrossRef CAS PubMed.
- T. A. Su, H. Li, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Nat. Chem., 2015, 7, 215–220 CrossRef CAS PubMed.
- M. L. Perrin, R. Frisenda, M. Koole, J. S. Seldenthuis, J. A. C. Gil, H. Valkenier, J. C. Hummelen, N. Renaud, F. C. Grozema, J. M. Thijssen, D. Dulić and H. S. J. van der Zant, Nat. Nanotechnol., 2014, 9, 830–834 CrossRef CAS PubMed.
- V. Kaliginedi, A. V. Rudnev, P. Moreno-Garcia, M. Baghernejad, C. Huang, W. Hong and T. Wandlowski, Phys. Chem. Chem. Phys., 2014, 16, 23529–23539 RSC.
- M. Baghbanzadeh, F. C. Simeone, C. M. Bowers, K.-C. Liao, M. Thuo, M. Baghbanzadeh, M. S. Miller, T. B. Carmichael and G. M. Whitesides, J. Am. Chem. Soc., 2014, 136, 16919–16925 CrossRef CAS PubMed.
- L. Jiang, C. S. S. Sangeeth and C. A. Nijhuis, J. Am. Chem. Soc., 2015, 137, 10659–10667 CrossRef CAS PubMed.
- A. Tan, J. Balachandran, B. D. Dunietz, S.-Y. Jang, V. Gavini and P. Reddy, Appl. Phys. Lett., 2012, 101, 243107 CrossRef.
- D. Fracasso, M. I. Muglali, M. Rohwerder, A. Terfort and R. C. Chiechi, J. Phys. Chem. C, 2013, 117, 11367–11376 CAS.
- A. Kovalchuk, T. Abu-Husein, D. Fracasso, D. A. Egger, E. Zojer, M. Zharnikov, A. Terfort and R. C. Chiechi, Chem. Sci., 2016, 7, 781–787 RSC.
- P. A. Van Hal, E. C. P. Smits, T. C. T. Geuns, H. B. Akkerman, B. C. De Brito, S. Perissinotto, G. Lanzani, A. J. Kronemeijer, V. Geskin, J. Cornil, P. W. M. Blom, B. De Boer and D. M. De Leeuw, Nat. Nanotechnol., 2008, 3, 749–754 CrossRef CAS PubMed.
- R. C. Chiechi, E. A. Weiss, M. D. Dickey and G. M. Whitesides, Angew. Chem., Int. Ed., 2008, 120, 148–150 CrossRef.
- G. Zhang, M. A. Ratner and M. G. Reuter, J. Phys. Chem. C, 2015, 119, 6254–6260 CAS.
- O. E. C. neda Ocampo, P. Gordiichuk, S. Catarci, D. A. Gautier, A. Herrmann and R. C. Chiechi, J. Am. Chem. Soc., 2015, 137, 8419–8427 CrossRef PubMed.
- P. I. Gordiichuk, G.-J. A. H. Wetzelaer, D. Rimmerman, A. Gruszka, J. W. de Vries, M. Saller, D. A. Gautier, S. Catarci, D. Pesce, S. Richter, P. W. M. Blom and A. Herrmann, Adv. Mater., 2014, 26, 4863–4869 CrossRef CAS PubMed.
- T. Abu-Husein, S. Schuster, D. A. Egger, M. Kind, T. Santowski, A. Wiesner, R. Chiechi, E. Zojer, A. Terfort and M. Zharnikov, Adv. Funct. Mater., 2015, 25, 3943–3957 CrossRef CAS.
- A. Natan, L. Kronik, H. Haick and R. Tung, Adv. Mater., 2007, 19, 4103–4117 CrossRef CAS.
- G. Heimel, F. Rissner and E. Zojer, Adv. Mater., 2010, 22, 2494–2513 CrossRef CAS PubMed.
- L. Yuan, R. Breuer, L. Jiang, M. Schmittel and C. A. Nijhuis, Nano Lett., 2015, 15, 5506–5512 CrossRef CAS PubMed.
- H. J. Yoon, K. C. Liao, M. R. Lockett, S. W. Kwok, M. Baghbanzadeh and G. M. Whitesides, J. Am. Chem. Soc., 2014, 136, 17155–17162 CrossRef CAS PubMed.
- D. A. Egger, F. Rissner, E. Zojer and G. Heimel, Adv. Mater., 2012, 24, 4403–4407 CrossRef CAS PubMed.
- V. Obersteiner, D. A. Egger, G. Heimel and E. Zojer, J. Phys. Chem. C, 2014, 118, 22395–22401 CAS.
- L. Cademartiri, M. M. Thuo, C. A. Nijhuis, W. F. Reus, S. Tricard, J. R. Barber, R. N. S. Sodhi, P. Brodersen, C. Kim, R. C. Chiechi and G. M. Whitesides, J. Phys. Chem. C, 2012, 116, 10848–10860 CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- A. Kokalj, J. Mol. Graphics Modell., 1999, 17, 176–179 CrossRef CAS PubMed.
- B. Hoeneisen, C. Mead and M.-A. Nicolet, Solid-State Electron., 1971, 14, 1057–1059 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10471a |
| This journal is © The Royal Society of Chemistry 2016 |