
Ti3C2Tx (MXene)–polyacrylamide nanocomposite films†
Michael Naguib*a,
Tomonori Saito*b,
Sophia Laib,
Matthew S. Ragerb,
Tolga Aytugb,
M. Parans Paranthaman*b,
Meng-Qiang Zhaoc and
Yury Gogotsic
aMaterials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: naguibma@ornl.gov
bChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: saitot@ornl.gov; paranthamanm@ornl.gov
cDepartment of Materials Science and Engineering, A.J. Drexel Nanomaterials Institute, Drexel University, Philadelphia, PA 19104, USA
First published on 20th July 2016
Abstract
Herein we report on the synthesis and characterization of MXene–polyacrylamide (PAM) nanocomposite films. Dimethylsulfoxide intercalation in-between the Ti3C2-based MXene layers led to full delamination of the MXene layers and hence a uniform dispersion of hydrophilic MXene nanosheets in aqueous PAM solutions was achieved. Polymer composite solutions of up to 75 wt% were synthesised. The as-prepared composite samples are flexible and the conductivity was increased significantly to 3.3 × 10−2 S m−1 with only 6 wt% (1.7 vol%) MXene loading.
A new large family of two dimensional (2D) transition metal carbides and carbonitrides, so called MXenes, has recently been developed.1,2 MXenes have a base composition of Mn+1Xn, where M is an early transition metal, X is either carbon or nitrogen, and n = 1–3.3 They are produced by selective etching of atomically thin metal layers from layered ceramics called MAX phases.4 Since MXenes are synthesized in fluoride containing aqueous solutions, their surfaces are terminated with a mixture of –O, –OH, and –F (hence after referred as Tx) groups.1,5 MXenes have a unique combination of properties, that includes electrical conductivity3,6,7 and hydrophilicity.3,8 They are also promising as electrode materials for Li- and Na-ion batteries9–12 and capacitors,13–15 and aqueous supercapacitors.8,16,17 In addition, they showed a great potential in other applications such as water purification18–20 and sensors.21,22 MXenes are considered to be excellent candidates for the production of functional nanocomposites due to their 2D morphology, hydrophilicity and fully functionalizable surfaces. However, attempts to incorporate MXenes in polymeric systems have been limited, only few studies have been reported thus far, for example by Ling et al.23 and Chen et al.24 In the former study, Ti3C2Tx MXene was mixed with either a charged polydiallyldimethylammonium chloride, PDDA, or an electrically neutral polyvinyl alcohol, PVA, to produce Ti3C2Tx/polymer composites.23 The Ti3C2Tx/PVA composite showed electrical conductivities as high as 2.2 × 104 S m−1, while the measured electrical conductivity for pure Ti3C2Tx was 2.4 × 105 S m−1. Furthermore, the tensile strength of the Ti3C2Tx/PVA composites was significantly enhanced compared to pure Ti3C2Tx or PVA film. It is worth noting that the smallest MXene loading reported by Ling et al. was 40 wt%, and since the film fabrication involved vacuum-assisted filtration of MXene/polymer solution, the MXene layers were highly oriented parallel to the film faces. More recently, Chen et al.24 grafted poly(2-(dimethylamino)ethylmethacrylate), PDMAEMA, brushes on 2D vanadium carbide, V2C, through self-initiated photo-grafting and photopolymerization, where dual-responsive properties of PDMAEMA as a function of environmental CO2 concentration and temperature have been investigated. In both cases, the key to intercalate MXenes in polymer matrices was the use of cationic-charged or hydrogen-bonding forming polymers to achieve a strong interaction with negatively charged OH/O/F terminated MXene surface.
To fabricate well-mixed MXene/polymer composites, one must also consider solubility in addition to polymer's favorable interaction with MXene. Dimethylsulfoxide, DMSO, is known to intercalate in-between the Ti3C2Tx layers causing an increase in the d-spacing.25 When the DMSO-intercalated Ti3C2Tx is immersed in water, a significant swelling in the structure takes place, leading to full delamination of the individual MXene layer. Since MXene has a highly negative zeta potential in neutral and basic solutions, once delaminated, it forms a stable colloidal solution in water.25,26
These two requirements motivated us to investigate polyacrylamide (PAM) as a candidate for the fabrication of MXene-based polymer composites. PAM is a water soluble, hydrogen-bond forming polymer, which is widely used in many industries such as in water and wastewater treatment processes, pulp and paper processing, mining and mineral processing, and as a soil conditioner or a solid support for the separation of proteins by electrophoresis.27,28 Several nanocomposites with PAM matrix have been reported. For example, Okay and Oppermann reported PAM–clay nanocomposite hydrogels by in situ polymerization of acrylamide with crosslinker in the presence of LAPONITE®,29 and Yang et al. reported multiwalled carbon nanotubes/PAM composites for Pb(II) and humic acid adsorption.30 Considering recent reports on MXene use for removal of lead and chromium ions from water,18,19 phosphate sequestration from wastewater,31 manufacturing biosensors21,32 and bactericidal films,33 incorporation of Ti3C2Tx into PAM becomes an important task.
Here we report on the fabrication of MXene/PAM nanocomposite films for the first time. Ti3C2Tx was synthesized and intercalated with dimethylsulfoxide (DMSO) and subsequently mixed with PAM in aqueous solutions. The rational behind using DMSO intercalated MXene as the starting material is the fact that DMSO intercalated MXene is known to spontaneously co-intercalate water between the MXene layers when exposed to humid air or water.25 Thus, once DMSO intercalated MXene is mixed with an aqueous solution of the polymer, it will spontaneously attract the solution between the layers leading to a well dispersed MXene/polymer composite. A series of MXene/PAM nanocomposites were cast from the aqueous solution mixture (Fig. 1) and their detailed microstructural and electrical property characterizations are presented.
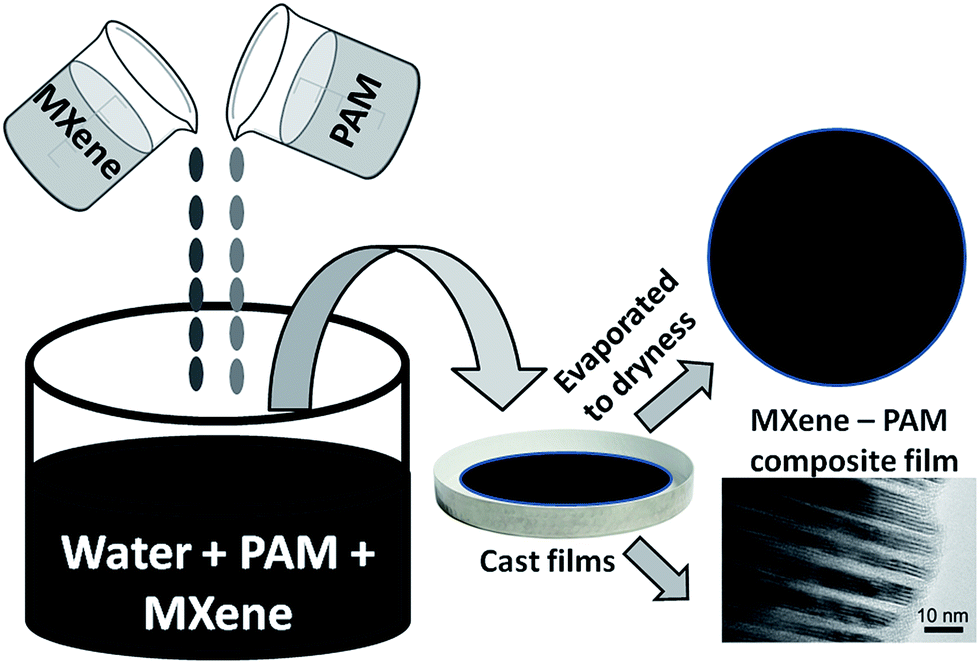 | ||
| Fig. 1 Schematic of the synthesis of MXene–PAM nanocomposite films. | ||
Ti3C2Tx was synthesized by etching Al from Ti3AlC2 using aqueous hydrofluoric acid, HF, (48%, Macron Fine Chemicals – Avantor Performance Materials, Center Valley, PA, USA) which was described in detail elsewhere.1–4 It is worth mentioning that glassware cannot be used with HF-based solutions and handling HF requires stringent safety precautions compared to other acids. The etching was achieved by soaking the Ti3AlC2 powder (−325 mesh) in the 48% HF solution with a ratio of 1 g powder: 10 mL solution, and stirred for 18 h at room temperature. This is followed by separation of powders from the solution by centrifuging, and washing using de-ionized (DI) water till a pH value higher than 4 is reached. The resulting slurry was then dried at room temperature using a vacuum-assisted filtration device for 48 h. The dried powders were then immersed in DMSO (99.9%, Fisher Scientific, China) with a ratio of 1 g MXene: 10 mL DMSO and stirred for 18 h at room temperature, followed by centrifuging to separate the excess DMSO from slurry before mixing with PAM, as described below.
PAM in 10 wt% aqueous solution was purchased from Polysciences. According to the manufacturer, the molecular weight of PAM is 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–1000000 g mol−1. DI water was purchased from BDH Chemicals and used without further purification. A schematic of the synthesis of MXene–PAM nanocomposite films is shown in Fig. 1. MXene–PAM composite films were prepared by varying the mass ratio of MXene to PAM. For instance, the preparation of 50 wt% (nominal) PAM–MXene composite film is described as follows: MXene (250 mg) in 4 mL DI water and PAM (250 mg) in 3 mL DI water were prepared in separate scintillation vials. Then each vial was vortexed for few seconds followed by sonication for 5 minutes. MXene/water mixture was poured into the PAM/water mixture, and 2 mL of DI water was used to rinse residual MXene into PAM-containing vial. The combined mixture was vortexed, then sonicated for 10–15 min until the solution attains visual homogeneity. Vortex mixing is performed right before casting the film, then the vial was gently swirled to get rid of bubbles. The PAM–MXene solution was poured into a Teflon tray, gently swirled for 1 min, covered to avoid dust contamination, and was let to dry at ambient temperature for 4–5 days. After this natural drying step, the film was further dried in an oven at 60 °C overnight.
000–1000000 g mol−1. DI water was purchased from BDH Chemicals and used without further purification. A schematic of the synthesis of MXene–PAM nanocomposite films is shown in Fig. 1. MXene–PAM composite films were prepared by varying the mass ratio of MXene to PAM. For instance, the preparation of 50 wt% (nominal) PAM–MXene composite film is described as follows: MXene (250 mg) in 4 mL DI water and PAM (250 mg) in 3 mL DI water were prepared in separate scintillation vials. Then each vial was vortexed for few seconds followed by sonication for 5 minutes. MXene/water mixture was poured into the PAM/water mixture, and 2 mL of DI water was used to rinse residual MXene into PAM-containing vial. The combined mixture was vortexed, then sonicated for 10–15 min until the solution attains visual homogeneity. Vortex mixing is performed right before casting the film, then the vial was gently swirled to get rid of bubbles. The PAM–MXene solution was poured into a Teflon tray, gently swirled for 1 min, covered to avoid dust contamination, and was let to dry at ambient temperature for 4–5 days. After this natural drying step, the film was further dried in an oven at 60 °C overnight.
To determine the MXene content in each composite sample, thermal gravimetric analysis, TGA, was conducted using a Q500 thermobalance from TA Instruments (New Castle, DE, USA). The samples were heated in platinum pan under an air flow of 35 mL min−1 from room temperature to 900 °C with a heating rate of 10 °C min−1. A Cu Kα (X1, Scintag, Cupertino, CA, USA) diffractometer operating at 45 kV and 40 mA was used to obtain the X-ray powder diffraction, XRD, for pristine MXene and MXene–PAM composites. The morphology of the MXene–PAM composites was investigated with scanning electron microscopy, SEM, (Zeiss Merlin VP, Carl Zeiss Microscopy GmbH, Oberkochen, Germany) and transmission electron microscopy, TEM, (JEOL JEM-2100, Japan) using an accelerating voltage of 200 kV. Cross-sections of the MXene–PAM films were produced by first embedding the films in epoxy resin and then cutting them using a glass microtome.
Resistance values were recorded with a digital multimeter (Hewlett Packard 34401A). To improve the electrical contact between the sample and the wire leads, a conductive silver epoxy was used. The temperature dependent resistance measurements were made in an environmental chamber (Tenney TJR) from room temperature to ∼70 °C.
MXene content in each MXene–PAM composite sample was determined from the TGA data shown in Fig. 2a and the data are summarized in Table 1. Heating the MXene in air results in its oxidation, forming titanium dioxide and carbon.34 The latter is then burned with further heating.35 An overall weight gain of ∼11% was observed from the TGA data of the pristine MXene paper (produced as described in ref. 23) which was used as the 100% MXene sample. Thus, the remaining weight percentage after heating each film was divided by 1.11 to estimate the actual percentage of MXene in the each sample. The total quantities of water and DMSO in these polymer MXene composite films are estimated to be in the range from 5.8–12.0 wt%, based on the initial weight loss in TGA below 220 °C. Although the drying step of the composite films were carefully conducted to remove DMSO, it is hard to estimate how much DMSO was still left. Once the film was exposed to moisture in the air, PAM tends to take up moisture from the air due to its hygroscopic nature. Thus, the solvent content observed in TGA is most likely water, but a little amount of DMSO may be remained in these composite films.
 | ||
| Fig. 2 (a) Thermogravimetric curves of MXene–PAM nanocomposite films with different actual Ti3C2Tx wt% and 100% MXene (“MXene paper”). (b) XRD patterns for Ti3C2Tx powders and PAM polymer in addition to MXene–PAM nanocomposites with different wt% of MXenes. Black squares represent the peaks of TiC secondary phase [PDF# 05-0693]. (c) and (d) SEM images for fracture surface of the 6 wt% MXene–PAM sample. | ||
| a Corrected for MXene wt gain due to oxidation.b Using the actual wt content and assuming densities of PAM and MXene to be 1.14 and 4.26 g cm−3, respectively. | ||||||
|---|---|---|---|---|---|---|
| Nominal wt% MXene | 0 | 10 | 50 | 70 | 80 | 100 |
| Remaining wt% from TGA | 1 | 7 | 34 | 69 | 83 | 111 |
| Actual MXene content, wt%a | NA | 6 | 31 | 63 | 75 | 100 |
| Estimated MXene content, vol%b | NA | 1.7 | 10.7 | 31.3 | 44.5 | 100 |
| Wt loss below 220 °C | 11.4 | 12.0 | 11.0 | 8.0 | 5.8 | 1.7 |
The XRD pattern of Ti3C2Tx (black curve in Fig. 2b) agrees well with the previously reported XRD patterns.1 Only small amount of TiC [PDF# 05-0693] from the MAX phase precursor was observed. As shown in Fig. 2b, a significant reduction in the intensity of the MXene (0002) peak around 9° 2θ was observed. This significant reduction indicates that most of the MXenes flakes are lacking order especially along the c-direction, which is indicative of well-dispersed flakes in the polymer. In fact, corroborating evidence for this comes from the SEM (Fig. 2c and d) and TEM (Fig. 3a and b) analysis where a good dispersion of the randomly oriented flakes is observed for the 6 wt% MXene film.
 | ||
| Fig. 3 TEM images of (a) and (b) 6 wt% MXene–PAM nanocomposite films supported on lacey carbon; and (c) and (d) 31 wt% MXene–PAM nanocomposite films. Dark lines in (b) and (d) correspond to single layers of MXene. | ||
The absence of ordered MXene stacks proves the efficacy for the design approach of using intercalated MXene where the intercalant is hygroscopic so it attracts the aqueous solution of the polymer and allows for an in situ polymerization between the MXene layers. In contrast, for the previously reported MXene–PVA or PDDA systems, the MXenes layers are found to be highly oriented,23 which is not desirable for applications where isotropic behavior is required. For the 6 wt% film, large quantities of PAM were intercalated into MXene layers, helping their separation. Therefore, well dispersed few-layer (<4) MXene nanosheets were present (Fig. 3a and b). With the reduction of PAM content (increase in MXene loading), the intercalation of PAM occurred every 3–8 MXene layers, which was not enough to fully delaminate the multi-layer MXenes and separate the MXene nanosheets. As a result, large amount of multi-layer MXene flakes, rather than few-layer flakes, were present in the 31 wt% film. These results indicate that the intercalation of PAM into MXene layers was a multi-stages process, which was similar to the process of intercalating chemical compounds into graphite.36
The conductivities of MXene–PAM nanocomposites were measured and are reported in Fig. 4. It should be noted that the nanocomposite films showed higher conductivity on the bottom side of the film due to the gravitational segregation of the MXene flakes during drying. Fig. 4a clearly shows that the room temperature electrical conductivity of the composite sample was significantly increased, by several orders of magnitude, to ∼3.3 × 10−2 S m−1 with as low as 1.7 vol% (6 wt%) MXene loading. Note that, due to its very high resistivity,37 our equipment capability was not enough to measure the room temperature electrical conductivity of pristine PAM and hence the value from ref. 37 has been used instead. With further increase in MXene loading, the conductivity of the nanocomposite films further increases, reaching to a value of 3.3 S m−1 at 44.5 vol% (75 wt%), denoting the formation of a more effective conductive network in the PAM matrix, where the conductivity is controlled by the MXene flakes.
 | ||
| Fig. 4 (a) Conductivity of various MXene–PAM nanocomposite films as a function of MXene volume loading. Measurements were taken on the more conductive side of the samples. Solid line represents the theoretical fit to the experimental data. Inset illustrates the power law dependence of conductivity above the percolation threshold, mth. (b) Temperature dependence of resistance for the 10.7 vol% (31 wt%) MXene–PAM nanocomposite film. | ||
As shown in Fig. 4, the increase in conductivity as a function of loading for the composite samples can be interpreted by a power-law expression
| σ ∝ (m − mth)γ for m > mth, |
The temperature dependent resistance for the 10.7 vol% (31 wt%) MXene–PAM composite film was measured and the data are presented in Fig. 4b. Note that the resistance of a material fluctuates with changes in temperature providing evidence of bulk properties. The sample displays a linear decrease in composite resistivity with increasing temperature, T, between 20 °C and 80 °C. This trend, negative temperature coefficient of resistance, TCR, effect, can be described using the following equation,

The observed decrease in resistance (increase in conductivity) with increasing the temperature may also be due to thermal expansion of PAM. Considering the residual moisture or DMSO (Fig. 2a) could plasticize PAM to lower its Tg.32 We see the composite films display a negative coefficient of resistance even at higher volume loadings thus showing the impact of the polymer matrix. We can conclude that MXene flakes are well encompassed by the polymer up to 1.7 vol% (6 wt%) and that little or no metallic contact is present. The composites up to this loading percentage exhibit unique single-sided, semiconductor-like properties. As the MXene loading is further increased to 10.73 vol% (31 wt%), the ratio of conducting-phase to polymer is likely high enough to allow a direct contact and form a percolation network, and the electrical properties gradually resemble a metal-like behaviour rather than semiconductor-like. This metal-like conductivity potentially allows the use of MXenes in radio frequency (RF) shielding and other applications, such as static charge dissipation, that require conductivity from polymers, especially in combination with improvement in mechanical properties. We observed an obvious increase in stiffness of the PAM after adding MXene, but detailed quantitative characterization of mechanical properties will be the subject of future studies. The most important outcome from this work is demonstration of a simple casting manufacturing process for a MXene-containing composite. With close to 20 different types of MXenes already reported and many coming, this offers an exciting opportunity for the development of a new generation of polymer-matrix composites containing MXenes.
In summary, we have successfully demonstrated the fabrication of MXene–PAM nanocomposite films. The presence of well dispersed nanoflakes of MXenes was confirmed by X-ray diffraction, SEM, and TEM. The TEM images indicate the presence of nanoflakes in 6 wt% MXene–PAM composite films. With increase in MXene loading, the conductivity increased significantly to 3.3 × 10−2 S m−1 for 6 wt% (1.7 vol%) MXene loaded films. With further increase in MXene loading, we observed non-uniformity between the top and bottom sides of the film, mainly due to the segregation of MXene flakes during drying. Improving methodology of MXene–PAM nanocomposite, especially at high MXene loading, will be a key for producing highly conductive nanocomposite films with desired functional properties. Among various polymer matrices tested, PAM showed a very promising dispersion of MXene nanoflakes, which indicates that using a hygroscopic intercalated MXene with a water soluble polymer can be an effective route to MXene–polymer composites. This work opens up various possibilities of MXene–polymer composites for many future applications.
Acknowledgements
This work was supported by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed by UT-Battelle, LLC, for the U.S. Department of Energy. Microscopy research was supported through a user proposal supported by ORNL's Center for Nanophase Materials Sciences (CNMS), which is sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. Synthesis of polymer composite research was supported (TS, SL, and MPP) by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division. YG sabbatical stay at ORNL was sponsored by the Fluid Interface Reactions, Structures and Transport (FIRST) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences.Notes and references
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- M. Naguib and Y. Gogotsi, Acc. Chem. Res., 2015, 48, 128–135 CrossRef CAS PubMed.
- H.-W. Wang, M. Naguib, K. Page, D. J. Wesolowski and Y. Gogotsi, Chem. Mater., 2016, 28, 349–359 CrossRef CAS.
- J. Halim, M. R. Lukatskaya, K. M. Cook, J. Lu, C. R. Smith, L.-Å. Näslund, S. J. May, L. Hultman, Y. Gogotsi, P. Eklund and M. W. Barsoum, Chem. Mater., 2014, 26, 2374–2381 CrossRef CAS PubMed.
- T. Hu, H. Zhang, J. Wang, Z. Li, M. Hu, J. Tan, P. Hou, F. Li and X. Wang, Sci. Rep., 2015, 5, 16329 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CAS.
- M. Naguib, J. Come, B. Dyatkin, V. Presser, P.-L. Taberna, P. Simon, M. W. Barsoum and Y. Gogotsi, Electrochem. Commun., 2012, 16, 61–64 CrossRef CAS.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, J. Am. Chem. Soc., 2013, 135, 15966–15969 CrossRef CAS PubMed.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- Y. Xie, Y. Dall’Agnese, M. Naguib, Y. Gogotsi, M. W. Barsoum, H. L. Zhuang and P. R. C. Kent, ACS Nano, 2014, 8, 9606–9615 CrossRef CAS PubMed.
- Y. Dall’Agnese, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Phys. Chem. Lett., 2015, 6, 2305–2309 CrossRef PubMed.
- J. Come, M. Naguib, P. Rozier, M. W. Barsoum, Y. Gogotsi, P.-L. Taberna, M. Morcrette and P. Simon, J. Electrochem. Soc., 2012, 159, A1368–A1373 CrossRef CAS.
- X. Wang, S. Kajiyama, H. Iinuma, E. Hosono, S. Oro, I. Moriguchi, M. Okubo and A. Yamada, Nat. Commun., 2015, 6, 6544 CrossRef CAS PubMed.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall’Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- R. B. Rakhi, B. Ahmed, M. N. Hedhili, D. H. Anjum and H. N. Alshareef, Chem. Mater., 2015, 27, 5314–5323 CrossRef CAS.
- Y. Ying, Y. Liu, X. Wang, Y. Mao, W. Cao, P. Hu and X. Peng, ACS Appl. Mater. Interfaces, 2015, 7, 1795–1803 CAS.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef CAS PubMed.
- C. E. Ren, K. B. Hatzell, M. Alhabeb, Z. Ling, K. A. Mahmoud and Y. Gogotsi, J. Phys. Chem. Lett., 2015, 6, 4026–4031 CrossRef CAS PubMed.
- F. Wang, C. Yang, C. Duan, D. Xiao, Y. Tang and J. Zhu, J. Electrochem. Soc., 2015, 162, B16–B21 CrossRef CAS.
- H. Liu, C. Duan, C. Yang, W. Shen, F. Wang and Z. Zhu, Sens. Actuators, B, 2015, 218, 60–66 CrossRef CAS.
- Z. Ling, C. E. Ren, M. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676–16681 CrossRef CAS PubMed.
- J. Chen, K. Chen, D. Tong, Y. Huang, J. Zhang, J. Xue, Q. Huang and T. Chen, Chem. Commun., 2015, 51, 314–317 RSC.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall'Agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, Nat. Commun., 2013, 4, 1716 CrossRef PubMed.
- M. Naguib, R. R. Unocic, B. L. Armstrong and J. Nanda, Dalton Trans., 2015, 44, 9353–9358 RSC.
- M. Friedman, J. Agric. Food Chem., 2003, 51, 4504–4526 CrossRef CAS PubMed.
- D. Taeymans, J. Wood, P. Ashby, I. Blank, A. Studer, R. Stadler, P. Gonde, P. Eijck, S. Lalljie, H. Lingnert, M. Lindblom, R. Matissek, D. Mueller, D. Tallmadge, J. O'Brien, S. Thompson, D. Silvani and T. Whitmore, Crit. Rev. Food Sci. Nutr., 2004, 44, 323–347 CrossRef CAS PubMed.
- O. Okay and W. Oppermann, Macromolecules, 2007, 40, 3378–3387 CrossRef CAS.
- S. B. Yang, J. Hu, C. L. Chen, D. D. Shao and X. K. Wang, Environ. Sci. Technol., 2011, 45, 3621–3627 CrossRef CAS PubMed.
- Q. Zhang, J. Teng, G. Zou, Q. Peng, Q. Du, T. Jiao and J. Xiang, Nanoscale, 2016, 8, 7085–7093 RSC.
- B. Xu, M. Zhu, W. Zhang, X. Zhen, Z. Pei, Q. Xue, C. Zhi and P. Shi, Adv. Mater., 2016, 28, 3333–3339 CrossRef CAS PubMed.
- K. Rasool, M. Helal, A. Ali, C. E. Ren, Y. Gogotsi and K. A. Mahmoud, ACS Nano, 2016, 10, 3674–3684 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, M. R. Lukatskaya, B. Dyatkin, C. Zhang, V. Presser, Y. Gogotsi and M. W. Barsoum, Chem. Commun., 2014, 50, 7420–7423 RSC.
- Z. Li, L. Wang, D. Sun, Y. Zhang, B. Liu, Q. Hu and A. Zhou, Mater. Sci. Eng., B, 2015, 191, 33–40 CrossRef CAS.
- H. Shioyama, Synth. Met., 2000, 114, 1–15 CrossRef CAS.
- S. Awasthi, K. Awasthi, R. Kumar and O. N. Srivastava, J. Nanosci. Nanotechnol., 2009, 9, 5455–5460 CrossRef CAS PubMed.
- S. Ansari and E. P. Giannelis, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 888–897 CrossRef CAS.
Footnote |
| † This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://www.energy.gov/downloads/doe-public-access-plan). |
| This journal is © The Royal Society of Chemistry 2016 |