
Theoretical insights into the alkaline metal M (M = Na and Cs) promotion mechanism for CO2 activation on the Cu(111) surface†
Abstract
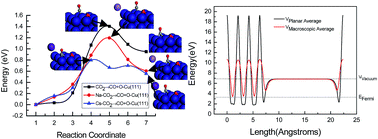
A systematic study on the alkaline metal M (M = Na and Cs) promotion mechanism for CO2 activation on the Cu(111) surface is presented for the first time based on self-consistent density functional theory calculations. The results show that CO2 adsorbs weakly and molecularly on a clean Cu(111) surface, whereas a drastic influence on the bonding, structure, and reactivity of adsorbed CO2 through the formation of CO2δ− radical anions was exerted by the presence of alkaline metals Na and Cs adatoms on the Cu(111) surface. This is the main physical origin of alkaline metals Na and Cs promotion in CO2 activation. The Na and Cs adatoms lower the dissociation activation barrier of CO2, in which the effect of Cs on CO2 dissociation is significantly larger than that of Na. Thus, the promotion effect of alkaline metals for the CO2 reduction into hydrocarbons on Cu catalysts can be attributed to a reduction of the dissociation activation barrier of CO2. The results also show that the origin of this promotion effect is predominately a direct electronic interaction between the Na and Cs-promoted Cu(111) surface and CO2 molecules. The presence of Na and Cs on the Cu(111) resulted in a decrease in the work function of the surface. The reduced amount of the work function on the Cs-promoted Cu(111) surface is more than that on the Na-promoted Cu(111) surface, explaining why the dissociation activation barrier of CO2 is lower on the Cs-promoted Cu(111) surface. The strong work function decrease of Na and Cs-promoted Cu(111) surface is corroborated by the presence of charge transfer. The charge transfer leads to the formation of a partially negative species, CO2δ−, which can be ascribed to the enhancement of back-donation of electrons from Na and Cs-promoted Cu(111) surface into an empty π orbital of CO2 compared to that on the clean Cu(111) surface. However, the back-donation mechanism of electrons is different between the Na and Cs-promoted Cu(111) surfaces, in which Na is an effective electron donor, whereas Cs is an electron acceptor, thus leading to the difference between promoting mechanisms of alkaline metals Na and Cs on CO2 activation. Cu as an electron donor in the Cs-promoted Cu(111) surface may result in a more reduced amount of the work function of Cs-promoted Cu(111) surface.
 Please wait while we load your content...
Please wait while we load your content...

