
Stripping voltammetric detection of copper ions using carbon paste electrode modified with aza-crown ether capped gold nanoparticles and reduced graphene oxide†
M. Ghanei-Motlagh*a,
Ch. Karamib,
M. A. Taherc and
S. J. Hosseini-Nasabd
aYoung Researchers and Elite Club, Kerman Branch, Islamic Azad University, Kerman, Iran. E-mail: m.ghaneimotlagh@yahoo.com
bDepartment of Chemistry, Kermanshah Branch, Islamic Azad University, Kermanshah, Iran
cDepartment of Chemistry, Faculty of Sciences, Shahid Bahonar University of Kerman, Kerman, Iran
dDepartment of Chemistry, Science and Research Branch, Islamic Azad University, Tehran, Iran
First published on 9th September 2016
Abstract
Based on the unique electronic properties of reduced graphene oxide (RGO) as well as the selective interaction between kryptofix 21-capped gold nanoparticles (GNPs) with copper(II) ions, a novel electrochemical sensor for the detection of Cu(II) ions in aqueous solutions was proposed. The best voltammetric response for Cu(II) was obtained with a paste composition of 61% (w/w) of graphite powder, 4.0% (w/w) of kryptofix 21-capped GNPs, 5.0% (w/w) of RGO and 30% (w/w) of paraffin oil using a solution of 0.1 mol L−1 HCl, an accumulation potential of −0.4 V vs. Ag/AgCl (3.0 mol L−1 KCl) and accumulation time of 180 s. Under these optimum experimental conditions, a sensitive response to Cu(II) ions within a wide concentration range (0.5–75 μg L−1) was obtained. The proposed electrochemical sensor showed a low detection limit (0.1 μg L−1), below the guideline value from the World Health Organization (WHO) and the United States Environmental Protection Agency (USEPA). The modified electrode was successfully utilized for the determination of Cu(II) in different water samples, and good recoveries were obtained.
1. Introduction
Copper is one of the most important elements in environmental, industrial, biological systems and in living organisms where it serves as a cofactor in at least 30 important enzymes.1 Exposure to copper manifests certain diseases in humans, for example, Menke's syndrome and Wilson's disease.2,3 However, reactivity and biological uptake of copper are strongly influenced by its free ion concentration.4 From this point of view, the quantitative determination of copper(II) in environmental samples is necessary. Several analytical methods have been successfully proposed for the determination of copper in environmental samples such as atomic absorption spectrometry (AAS),5 fluorescence spectrometry,6 inductively coupled plasma optical emission spectrometry (ICP-OES),7 inductively coupled plasma mass spectrometry (ICP-MS),8 graphite furnace atomic absorption spectrometry (GFAAS)9 and different electrochemical methods.10–12 Among the aforementioned methods, electrochemical methods are the most favorable techniques for the determination of metal ions because of their low cost, high sensitivity, experimental simplicity, low sample volume and the ability for portability.13–16In the modern electroanalytical analysis, the key factor is electrode modification, which requires the selection of suitable material to improve the electrochemical performance.17,18 The self-assemble monolayer (SAM) provides a simple method of electrode functionalization by strong chemisorptions.19 Modification of the gold electrodes surface via functionalization of thiol thin film SAMs by using organic functional groups such as aldehydes, amines, amides, and carboxylic acids are attractive and of prime importance in sensor technology.20 Notable studies were published by Wei and co-workers, who applied a self-assembled monolayer of azacrown ether derivative as Cr(VI) complexing agents on a gold electrode.21 Shervedani and Mozaffari described devices based on gold cysteamine SAM functionalized with salicylaldehyde, which resulted in the effective determination of Cu(II) in blood serum samples by Osteryoung square wave voltammetry (OSWV) and electrochemical impedance spectroscopy (EIS).22 The proposed electrochemical sensor not only exhibited good reproducibility, but also showed a low detection limit.
In particular, gold nanoparticles (GNPs) have demonstrated to be very appropriate for the development of modified electrodes and allow the construction of electrode nanoarrays in one or several dimensions. Furthermore, the use of GNPs in electrochemical studies have received considerable attention due to their relatively large surface area, high conductivity, suitable electro-catalysis characteristics, good biocompatibility, and their interface-dominant properties, which significantly differ from those of their bulk counterparts.23 Colloidal gold-modified electrodes have been prepared using glassy carbon,24 ultra-microelectrode arrays,25 metal Au26 and carbon paste27 as electrode substrates.
Recently, graphene has received extensive attention due to its unique electronic, thermal, and mechanical features.28 The surface of graphene oxide (GO) bears different oxygen-containing functional groups such as carboxyl, hydroxyl, epoxy, and keto groups.29 The presence of these functional groups destroys the planar sp2 carbons of graphite and converts it to sp3 carbons.30 Therefore, the π–π electronic conjugation of graphite is destroyed in GO, resulting in a significant decrease in electrical conductivity.31 Hence, a reduction process should be accompanied to improve the electrical conductivity of GO. This approach can successfully remove oxygen functionalities from the surface of GO and produce reduced graphene oxide (RGO). RGO possesses a higher conductivity compared with GO32 which holds a great promise for electrochemical sensing applications.
The development of selective electrochemical sensor has received widespread attention during the past three decades because of their possibility use in complex real samples, as they provide a rapid, accurate and low price method of analysis. The essential requirements to impart selectivity of electrochemical sensor are to use specific ion-recognition elements which show a strong affinity for the target ion. Macrocyclic compounds, due to their selective receptor properties and ease of structural modification, have been employed as modifier agents in the construction of the modified electrode for the determination of different metal ions.33–37 Aza-crown ether macrocycles serve as simple receptor models and are well-suited as hosts because of their conformational flexibility and the presence of multiple binding sites.38–40 Although these compounds exhibit elevated selectivity to certain metal ions, the use of them in the volt-amperometric analysis has received little attention, probably because of their nonconductive property.
The main goal of this investigation is to develop a novel copper(II) electrochemical sensor based on kryptofix 21 functionalized GNPs and RGO. The crucial parameters, linear response range, and detection limit of the proposed sensor in the detection of Cu(II) were examined. The resulting modified electrode has also been employed for voltammetric detection of copper ions in different water samples.
2. Experimental
2.1. Materials and instrumentation
Chemicals including tetrachloroauric(III) acid (HAuCl4·3H2O), 11-mercaptoundecanoic acid (MUA) and dodecanethiol were obtained from Sigma-Aldrich (St. Louis, MO, USA). Sodium borohydride (NaBH4), highly pure graphite powder (particle size <50 μm, electrical conductivity 2 × 104 S m−2), high viscosity paraffin oil (density = 0.88 kg L−1), triflic anhydride ((CF3SO2)2O) and 1,4,10-trioxa-7,13-diazacyclopentadecane (kryptofix 21) were purchased from Merck (Darmstadt, Germany). The copper stock solution in 0.1% nitric acid was supplied by Sigma-Aldrich. All other reagents were acquired from standard sources and used as received.A Cary 50 single detector double beam in-time spectrophotometer (Varian, Australia) was used for recording the absorption spectra. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images were taken with a Philips CM 200 TEM operating at 200 kV and Hitachi S 4160 SEM operating at 30 kV respectively. The Fourier transform infrared (FT-IR) spectra were recorded in the range of 4000–400 cm−1 on a Spectrum One FT-IR spectrometer (PerkinElmer, Norwalk, CT, USA). Differential pulse voltammetry (DPV) measurements were performed using an AUTOLAB TYPE III (Eco-Chemie B.V., Utrecht, The Netherlands) potentiostat. A typical three-electrode system consisted of a modified carbon paste electrode (MCPE) as working electrode, an Orion Ag/AgCl double-junction reference electrode and a platinum wire counter electrode. Electrochemical measurements were conducted at room temperature (25 ± 1 °C) under a purified nitrogen atmosphere. All pH values of solutions were determined by a Metrohm digital pH-meter model 827 (Herisau, Switzerland). Ultra-pure water (18 mΩ) was obtained from a Milli-Q water purification system (Millipore Corporation, Milford, MA, USA). The containers and glassware used in the electrochemical studies were soaked in 3 mol L−1 nitric acid overnight and rinsed with deionized water before use.
2.2. Preparation of MUA-capped GNPs
The starting monolayer-protected nanoparticles with dodecanethiol were prepared and purified by the reduction of Au(III) ions with NaBH4 according to the Brust method.41 For the synthesis of MUA-capped GNPs, the place exchange reaction was applied according to methods elsewhere described.42,43 In brief, the MUA-capped GNPs was prepared by dissolving (50 mg, 0.06 mmol) of the dodecanethiol-capped GNPs in 10 mL tetrahydrofuran and then (40 mg, 0.18 mmol) of 11-mercapto undecanoic acid was added and the mixture was stirred at room temperature for 6 hours. The resulting dark brown solution was concentrated and washed successively with ethanol.2.3. Preparation of the kryptofix 21-capped GNPs
The kryptofix 21-capped GNPs was prepared by dissolving (40 mg, 0.05 mmol) of the MUA-capped GNPs in 10 mL dichloromethane and then 70 mg (0.25 mmol) of triflic anhydride was added under vigorous stirring at room temperature for 10 min. Then, 65 mg (0.24 mmol) of kryptofix 21 was added to the mixture with vigorous stirring at room temperature for 30 min. The solvent was removed via rotary evaporation and the obtained dark brown precipitate washed successively with deionized water and acetone (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, v/v) to remove the unreacted materials. A schematic diagram for an entire process of the kryptofix 21-capped GNPs preparation is shown in Fig. 1.
:7, v/v) to remove the unreacted materials. A schematic diagram for an entire process of the kryptofix 21-capped GNPs preparation is shown in Fig. 1.
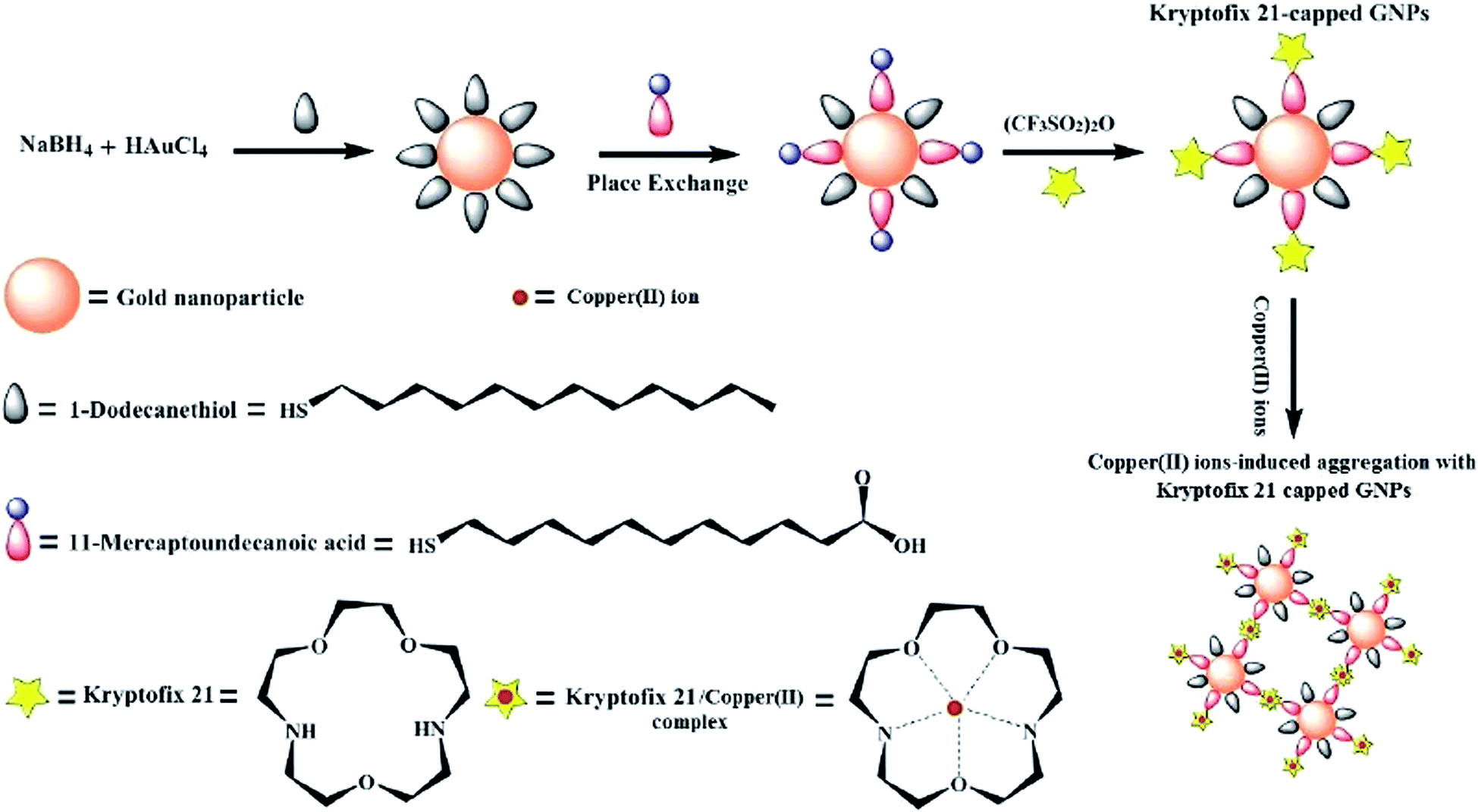 | ||
| Fig. 1 A schematic diagram for synthesis of kryptofix 21-capped GNPs. | ||
2.4. Preparation of RGO
GO was prepared using graphite powders according to the procedure offered in the literature.44 RGO was prepared by reduction of GO using hydrazine and ammonia solutions. To synthesize RGO, 0.1 g of GO was added to 150 mL of water, followed by sonicating for 2 h. Then, 0.1 mL of ammonia (28% w/w) and 1.4 mL of hydrazine (80% w/w) were added to the GO resultant dispersion. After being vigorously stirred for a few minutes, the reaction mixture was stirred for 1 h at 95 °C. After this, the mixture turned its color from brown to black, which indicated the reduction of GO to RGO. Consequently, this mixture was centrifuged, and the prepared RGO was washed with water and then dried at 70 °C in vacuum for 16 h.2.5. Preparation of MCPE
The modified carbon paste was prepared by mixing a known quantity of graphite powder, kryptofix 21-capped GNPs and RGO with paraffin oil using a mortar and pestle and was allowed to homogenize for 24 h. The MCPE was then fabricated by packing the prepared paste into a Teflon tube (2 mm inner diameter and 10 cm long). A copper wire was dissected through the paste, to provide an electrical contact. The external surface of MCPE was smoothed on a weighing paper. Fresh electrode surfaces were produced by scraping out of the surface with a spatula and replaced by modified paste and polishing it against weighing paper until the surface had a shiny appearance.2.6. Analytical procedure
Detection of Cu(II) with proposed electrochemical sensor was evaluated by differential pulse anodic stripping voltammetry (DPASV). The cell containing the target analyte ion at a given concentration in 0.1 M HCl solution (20 mL) was subjected to electrolysis under stirring at the potential of −0.4 V. After 3 min electrolysis, the voltammetric monitoring was performed in the differential pulse anodic stripping mode of operation from −500 to +200 mV (versus Ag/AgCl, 3.0 mol L−1 KCl) at scan rate of 50 mV s−1.3. Results and discussion
3.1. Characterization
The (FT-IR) spectra for MUA-capped GNPs and kryptofix 21-capped GNPs are shown in Fig. 2. According to the FT-IR spectrum of the MUA-capped GNPs (Fig. 2A), the band at 1704 cm−1 is due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of carboxylic acid groups. The absorption peaks at 2847 cm−1 and 2918 cm−1 are attributed to the stretching vibrations of C–H. The broadband at 3444 cm−1 is related to O–H stretching vibration of carboxylic acid groups in the MUA-capped GNPs. In kryptofix 21-capped GNPs spectrum (Fig. 2B), the absorption peaks appearing at 1513 and 3700–3500 cm−1 are corresponded to the amide groups existing in the prepared kryptofix 21-capped GNPs. These results implying that the kryptofix 21 molecules were successfully linked on the surface of MUA-capped GNPs.
O stretching vibration of carboxylic acid groups. The absorption peaks at 2847 cm−1 and 2918 cm−1 are attributed to the stretching vibrations of C–H. The broadband at 3444 cm−1 is related to O–H stretching vibration of carboxylic acid groups in the MUA-capped GNPs. In kryptofix 21-capped GNPs spectrum (Fig. 2B), the absorption peaks appearing at 1513 and 3700–3500 cm−1 are corresponded to the amide groups existing in the prepared kryptofix 21-capped GNPs. These results implying that the kryptofix 21 molecules were successfully linked on the surface of MUA-capped GNPs.
 | ||
| Fig. 2 FT-IR spectra of (A) MUA-capped GNPs and (B) kryptofix 21-capped GNPs. | ||
The TEM images of the kryptofix 21-capped GNPs before and after additions of copper ions are shown in Fig. 3. As can be seen, the kryptofix 21-capped GNPs are well dispersed with spherical shape in the absent of analyte ions. Also, the average particle size of kryptofix 21-capped GNPs were around 1.5 ± 0.5 nm. Upon adding of copper(II) ions, the TEM image of the kryptofix 21-capped GNPs is exhibited a significant aggregation due to a sandwich complex between kryptofix 21 moiety and Cu(II) ions.
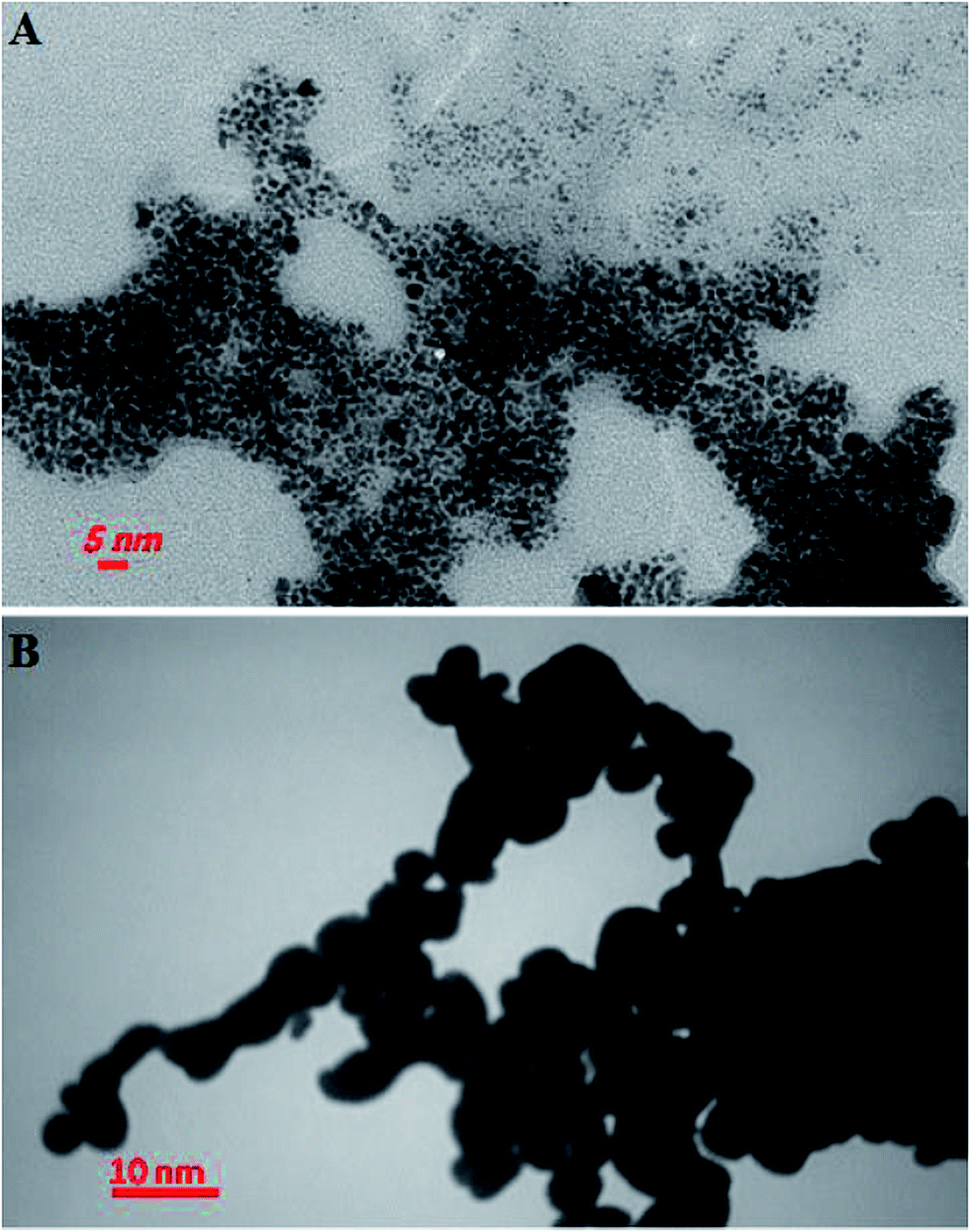 | ||
| Fig. 3 Typical TEM image of (A) kryptofix 21-capped GNPs and (B) kryptofix 21-capped GNPs in the presence of 0.1 mmol L−1 copper(II) ions. | ||
To characterize the prepared RGO, different techniques involving FT-IR, FE-SEM and TEM were also conducted. As shown in Fig. S1(A),† the characteristic peaks of CO and O–H groups were at 3373 cm−1 and 1724 cm−1 in GO, respectively. Compared with GO (Fig. S1(B)†), the peak intensity at about 3373 cm−1 for O–H groups decreases significantly, and the peak at 1724 cm−1 disappears indicating the successful reduction of GO.
Moreover, the FE-SEM and TEM images of RGO are shown in Fig. S2.† As can be seen from Fig. S2(A),† the RGO shows layer-like structure with a fairly smooth surface. As observed in TEM image (Fig. S2(B)†), free-standing RGO sheet exhibits ultra-thin, smooth and un-wrinkled structure.
3.2. Preliminary study
Before the application of the aza-crown ether capped GNPs in the electrochemical detection of Cu(II) ions, the sensing ability of the modified GNPs with various metal ions in tetrahydrofuran/H2O (3:7, v/v) solutions was investigated. As can be seen from Fig. 4, absorbance spectrum of modified GNPs has no surface plasmon resonance (SPR) around 520 nm and hence, reveals that the kryptofix 21-capped GNPs have average core diameter less than 2 nm.45 Furthermore, only upon incubation with Cu(II) ion, a pink color was monitored, and a characteristic absorption at about 538 nm was observed. From above result, it is immediately possible to distinguish the selective interaction of copper ions with the prepared modified GNPs.
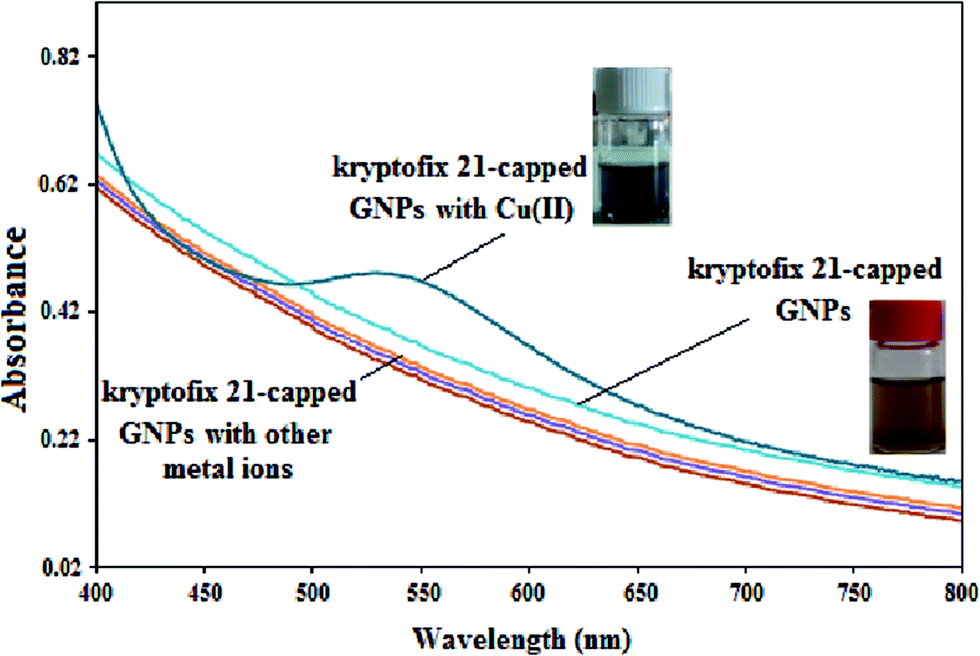 | ||
| Fig. 4 Absorption spectra of the kryptofix 21-capped GNPs in the presence of various metal ions (0.1 mmol L−1) such as Fe(III), Hg(II), Ag(I), Al(III), Ca(II), Cd(II), Co(II), Cr(III), Mn(II), Ni(II), Zn(II), Pb(II) and Cu(II). | ||
3.3. Anodic stripping voltammetric behavior of Cu(II) on MCPE
Preliminary experiments were carried out to identify the general features, which characterize the electrochemistry of Cu(II) ions at MCPEs surface. Fig. 5 shows the typical DPV curves obtained using unmodified CPE (A), RGO–CPE (B), kryptofix 21-capped GNPs–CPE (C) and RGO/kryptofix 21-capped GNPs–CPE (D) after preconcentrating in 15 μg L−1 Cu(II) solution. As can be seen, the observed electroanalytical signals increased in the following order: CPE < MCPE with RGO < MCPE with kryptofix 21-capped GNPs < MCPE with RGO/kryptofix 21-capped GNPs. From these results, it can be suggested that the modification of CPE with RGO and kryptofix 21-capped GNPs enhance copper binding sites and electrical conductivity of the proposed electrode, providing an advantageous platform for sensing applications. Moreover, only a residual current was observed when RGO/kryptofix 21-capped GNPs–MCPE was applied in the absence of Cu(II) ions (Fig. 5E). | ||
| Fig. 5 Differential pulse voltammograms of (A) unmodified CPE, (B) RGO–CPE, (C) kryptofix 21-capped GNPs-CPE, (D) RGO/kryptofix 21-capped GNPs–CPE, with Cu(II) concentration of 15 μg L−1 and (E) RGO/kryptofix 21-capped GNPs–CPE in the absence of Cu(II) ions. Other conditions; stripping solution = 0.1 mol L−1 HCl, accumulation potential = −0.4 V (vs. Ag/AgCl), accumulation time = 180 s, scan starting from −0.5 to +0.2 V, scan rate = 50 mV s−1 and pulse period = 4 ms. Carbon paste composition; of 60% (w/w) of graphite powder, 5.0% (w/w) of kryptofix 21-capped GNPs, 5.0% (w/w) of RGO and 30% (w/w) of paraffin oil. | ||
3.4. Optimization of significant parameters
As mentioned above, the voltammetric peak current of copper(II) ions is heightened at the surface of the proposed RGO/kryptofix 21-capped GNPs electrode. However, optimization of some important experimental parameters for the determination of Cu(II) is of critical importance. Hence, the effect of the electrode composition, pH of the medium, supporting electrolyte concentration, accumulation time and accumulation potential were conducted and optimized in copper(II) solution of 15 μg L−1.The influence of variation in the kryptofix 21-capped GNPs present in the composition of MCPE was initially evaluated (Fig. 6). After changing the mediator present from 1.0 to 7.0% (w/w), the signal of Cu(II) increased markedly, probably due to the increase of accumulation ability. The peak current decreased significantly when more than 4.0% w/w of kryptofix 21-capped GNPs is used in the MCPE composition. It can be ascribed to the decrease in the conductive area at the surface of MCPE. Therefore, 4.0% w/w of kryptofix 21-capped GNPs in paste composition of MCPE was applied for all the subsequent measurements.
 | ||
| Fig. 6 Anodic peak currents obtained by DPASV as function of the kryptofix 21-capped GNPs percentages in MCPE for 15 μg L−1 Cu(II) solution. Other conditions were the same as in Fig. 5. | ||
The percentage of RGO in carbon paste composition was also investigated. The stripping peak current was found to increase with increasing the RGO percentage up to 5.0% w/w and leveled off at a higher percentage. According to experimental results a carbon-paste composition of 4.0% kryptofix 21-capped GNPs, 5.0% RGO, 61% graphite and 30% (w/w) paraffin oil was used as optimal carbon paste composition in this work.
The influence of the solution pH on the electrochemical response of the proposed MCPE toward Cu(II) detection was evaluated by DPASV. The variations of the stripping peak current with respect to the change of solution pH are shown in Fig. 7. Considering the fact that the solution pH and ion strength are dependent on the concentration of HCl solution. Therefore, 0.1 mol L−1 HCl (pH = 1) was selected for preconcentration solutions in the following assays.
 | ||
| Fig. 7 Study of the anodic peak currents obtained by DPASV as function of the preconcentration solution pH in the presence of 15 μg L−1 Cu(II). Other conditions were the same as in Fig. 5. | ||
The effect of HCl concentration on the electroanalytical response of 15 μg L−1 Cu(II) was investigated. The voltammetric peaks were obtained in 0.05, 0.1, 0.15, 0.25 and 0.5 mol L−1 of HCl, using the following procedure: accumulation potential = −0.4 V (vs. Ag/AgCl), accumulation time = 180 s, scan starting from −0.5 to +0.2 V, scan rate = 50 mV s−1 and pulse period = 4 ms. The maximum peak currents were achieved at 0.1 mol L−1 HCl and therefore this concentration was chosen as supporting electrolyte for further experiments.
The dependence of accumulation potential on the stripping peak current was studied. For this purpose, the voltammetric measurements of a solution containing 15 μg L−1 of analyte ion solution were evaluated using accumulation potential values of −0.1, −0.2, −0.3, −0.4, −0.5, −0.6, −0.7 and −0.8 V in 0.1 mol L−1 HCl solution and applying an accumulation time of 180 s. It was observed that reduction potentials less negative than −0.4 V vs. Ag/AgCl reference electrode produce an analytical signal with low intensity and, consequently, inappropriate for Cu(II) detection. Moreover, the potentials more negative than −0.6 V led to decreased peak currents. Hence, for further study the accumulation potential of −0.4 V vs. Ag/AgCl reference electrode was employed, since more negative potential could negatively affect the selectivity of the proposed MCPE.
Because the copper(II) voltammetric signals was also affected by accumulation time, it was necessary to find an optimum value of the accumulation time for efficient stripping of analyte ions on the surface of MCPE. Evaluation of the accumulation time unveiled almost linear increase of the Cu(II) response up to 180 s followed by leveling off at higher accumulation times as a consequence of surface saturation. Therefore, reduction time of 180 s was applied throughout, as it combines good sensitivity and relatively short analysis time.
3.5. Study of interferences
The interference studies were carried out by evaluating the interference effects of K(I), Na(I), Ca(II), Cd(II), Ni(II), Mn(II), Zn(II), Hg(II), Pb(II), Al(III) and Cr(III) under the optimum conditions. The tolerance level was defined as the maximum amount of interfering ions can produce an error of ±5% on the Cu(II) current.46 The selectivity of the MCPE was investigated by calculating the stripping peak current ratio (Is/I0), where Is and I0 were the peak current of Cu(II) in the present and absence of co-existing interference ions, respectively. As documented by Fig. 8, a 400-fold excess of Na(I), K(I), 300-fold excess of Ca(II), 250-fold excess of Al(III), 100-fold excess of Ni(II), Cd(II), Mn(II), Zn(II), 40-fold excess of Hg(II), Pb(II) and Cr(III) over copper(II) hardly causes the significant change of copper(II) voltammetric signals, in which signal current ratio only slightly varied from 0.96 to 1.04. Evidently, taking as the advantage of the selective complex forming ability of the kryptofix 21-capped GNPs with copper(II) ions, the use of the MCPE can add some discrimination power to resolve the metal ion mixture. | ||
| Fig. 8 The voltammetric peak current ratio (Is/I0) of Cu(II) (15 μg L−1) in the presence of possible interference ions; K(I) (6 mg L−1), Na(I) (6 mg L−1), Ca(II) (4.5 mg L−1), Al(III) (3.75 mg L−1), Ni(II) (1.5 mg L−1), Cd(II) (1.5 mg L−1), Mn(II) (1.5 mg L−1), Zn(II) (1.5 mg L−1), Hg(II) (600 μg L−1), Pb(II) (600 μg L−1) and Cr(III) (600 μg L−1). | ||
3.6. Analytical performance for the detection of Cu(II)
To provide more information about the electroanalytical performance, the calibration data for Cu(II) using MCPE under the optimal operational conditions (i.e., 0.1 mol L−1 HCl solution, 180 s accumulation time, and −0.4 V accumulation potential) were obtained. A representative curve is shown in Fig. 9. As can be seen, the DPASV responses of the MCPE varied linearly toward Cu(II) concentration at a wide concentration range of 0.5–75 μg L−1. The linearization equation was found to be IP (μA) = 0.665C (μg L−1) + 0.413 with a correlation coefficient (R2) of 0.999. The detection limit of 0.1 μg L−1 was obtained with the calculation based on the signal-to-noise characteristics of S/N = 3. The achieved detection limit is well below the guideline copper value in drinking water set by United States Environmental Protection Agency (USEPA, 1.0–2.0 μg L−1) and by World Health Organization (WHO, 1.3 μg L−1).47 The relative standard deviation (RSD) value for 15 μg L−1 Cu(II) from the seven parallel measurements was calculated as 3.1%. Table 1 summarizes the analytical characteristics of the current method with other electrochemical methods11,48–55 described in the literature for copper(II) detection. From several points of view, the comparison is favorable to the procedure here discussed.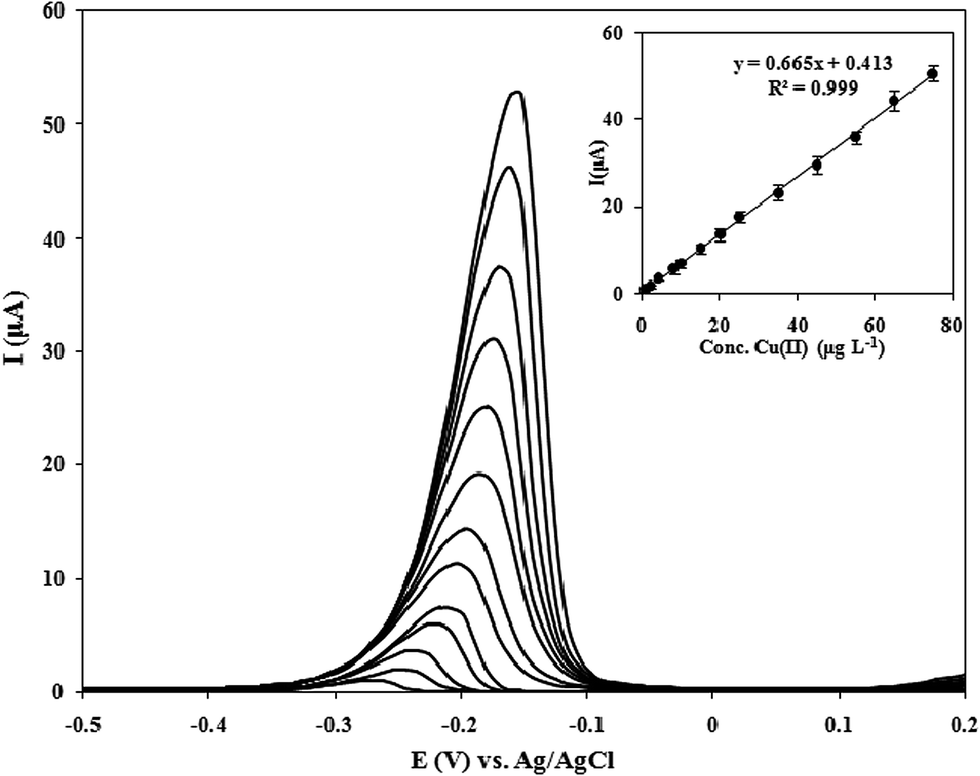 | ||
| Fig. 9 Differential pulse voltammograms with different concentrations of Cu(II) at optimal conditions. Inset shows the related calibration curve. | ||
| Modifiera | Electrode typeb | Analysis methodc | Detection limit (μg L−1) | Linear range (μg L−1) | Ref. |
|---|---|---|---|---|---|
| a IIP: ion imprinted polymer, MWCNTs: multiwalled carbon nanotubes, CTS-ECH: chitosan crosslinked with epichlorohydrin, APS: aminopropyl-grafted silica gel, Ac-Phos SAMMS: carbamoylphosphonic acid (acetamide phosphonic acid) self-assembled monolayer on mesoporous silica, MIPs: molecular imprinted polymers, SbF: antimony film, SiAt: 2-aminothiazole organofunctionalized silica, DMSA: meso-2,3-dimercaptosuccinic acid, NZ: natural zeolite, GNPs: gold nanoparticles, RGO: reduced graphene oxide.b CPE: carbon paste electrode, CNPE: carbon nanotubes paste electrode, SPCPE: solid paraffin-based carbon paste electrode, GE: gold electrode.c DPASV: differential pulse anodic stripping voltammetric, LSASV: linear scan anodic stripping voltammetry, SWV: square wave voltammetry, AdSV: adsorptive stripping voltammetry, DPV: differential pulse voltammetry, DPCSV: differential pulse cathodic stripping voltammetry. | |||||
| IIP/MWCNTs | CPE | DPASV | 0.34 | 2–120 | 11 |
| CTS-ECH | CNPE | LSASV | 0.6 | 5–1016 | 48 |
| APS | CPE | SWV | 0.19 | 3.1–12.7 | 49 |
| Ac-Phos SAMMS | CPE | AdSV | 0.5 | 10–200 | 50 |
| MIPs | CPE | DPV | 1.46 | 4.4–63.5 | 51 |
| SbF | CPE | SWV | 1.45 | 0–120 | 52 |
| SiAt | SPCPE | DPASV | 1.97 | 4.7–158.8 | 53 |
| DMSA | GE | DPASV | 1.29 | 3–225 | 54 |
| NZ | CPE | DPCSV | 0.95 | 3.2–317 | 55 |
| Kryptofix 21-capped GNPs/RGO | CPE | DPASV | 0.1 | 0.5–75 | This work |
3.7. Practical application
Different real water samples, including river water (Sarcheshmeh, Rafsanjan, Iran), Tap water (Sirjan drinking water, Kerman, Iran), well water (Shahid Bahonar University of Kerman, Kerman, Iran), aqueduct water (Balvard, Sirjan, Iran), and dam water (Chahar Gonbad, Sirjan, Iran) were selected to study the suitability of the proposed MCPE for the detection of Cu(II), as an example of its applicability for the analysis of environmental samples. Water samples were filtered through a 0.2 μm pore-size, polycarbonate membrane filters, to remove any suspended material. The content of copper was then determined by means of the standard addition method. The analytical results and recoveries are listed in Table 2. The recoveries of the spiked sample were evaluated using the t-test, and at 95% confidence level there is no significant difference between the results of the developed method and data obtained by GF-AAS as an independent technique. This result proves that the prepared electrochemical sensor has a proper capability for accurate and precise determination of copper in environmental water samples.| Sample | Voltammetry | Recoverya (%) ± RSDb | GF-AAS | |
|---|---|---|---|---|
| Added (μg L−1) | Found (μg L−1) | Found (μg L−1) | ||
| a Average value from three individual experiments.b Relative standard deviations (n = 3).c Not detected. | ||||
| River water | — | 5.7 | — | 6.0 |
| 10.0 | 15.4 | 98.0 ± 3.0 | — | |
| 15.0 | 21.3 | 102.9 ± 3.3 | — | |
| Tap water | — | 3.1 | — | 2.9 |
| 10.0 | 13.3 | 101.5 ± 2.4 | — | |
| 15.0 | 17.7 | 97.8 ± 3.1 | — | |
| Aqueduct water | — | N.D.c | — | N.D. |
| 10.0 | 9.8 | 98.0 ± 2.7 | — | |
| 15.0 | 14.5 | 96.6 ± 3.1 | — | |
| Dam water | — | N.D. | — | N.D. |
| 10.0 | 10.2 | 102.0 ± 2.8 | — | |
| 15.0 | 15.6 | 104.0 ± 2.5 | — | |
4. Conclusions
The modification of GNPs enables ready installation of a wide variety of functional interfaces, leading to excellent specificity and selectivity for any desired analyte when the materials are used as modifiers in electrochemical sensors. The kryptofix 21-capped GNPs provides a novel sensing material for the detection of copper(II) ions at ppb levels. The combination of kryptofix 21-capped GNPs with RGO offers significant electroanalytical performance. The suggested modified electrode had several advantages such as simple operation, low cost, remarkable selectivity, high sensitivity and good reproducibility. Under optimized conditions, selective detection of Cu(II) in a linear concentration range of 0.5–75 μg L−1 was obtained. It has also been shown that the prepared MCPE can be applied for trace copper determination in different water samples.References
- M. Ghanei-Motlagh, M. A. Taher, V. Saheb, M. Fayazi and I. Sheikhshoaie, Electrochim. Acta, 2011, 56, 5376–5385 CrossRef CAS.
- P. C. Bull and D. W. Cox, Trends Genet., 1994, 10, 246–252 CrossRef CAS PubMed.
- V. L. Hodgkinson, S. Zhu, Y. Wang, E. Ladomersky, K. Nickelson, G. A. Weisman, J. Lee, J. D. Gitlin and M. J. Petris, Am. J. Physiol.: Cell Physiol., 2015, 309, C660–C668 Search PubMed.
- M. H. Mashhadizadeh, S. Ramezani and S. Ebrahimi, Sens. Actuators, B, 2012, 169, 305–311 CrossRef CAS.
- M. Ghaedi, K. Niknam, K. Taheri, H. Hossainian and M. Soylak, Food Chem. Toxicol., 2010, 48, 891–897 CrossRef CAS PubMed.
- P. Xi, J. Dou, L. Huang, M. Xu, F. Chen, Y. Wu, D. Bai, W. Li and Z. Zeng, Sens. Actuators, B, 2010, 148, 337–341 CrossRef CAS.
- R. Gao, Z. Hu, X. Chang, Q. He, L. Zhang, Z. Tu and J. Shi, J. Hazard. Mater., 2009, 172, 324–329 CrossRef CAS PubMed.
- N. Zhang, H. Peng and B. Hu, Talanta, 2012, 94, 278–283 CrossRef CAS PubMed.
- M. N. Matos Reyes and R. C. Campos, Talanta, 2006, 70, 929–932 CrossRef PubMed.
- V. K. Gupta, R. N. Goyal, N. Bachheti, L. P. Singh and S. Agarwal, Talanta, 2005, 68, 193–197 CrossRef CAS PubMed.
- H. Ashkenani and M. A. Taher, J. Electroanal. Chem., 2012, 683, 80–87 CrossRef CAS.
- L. Zou, Y. Li, W. Zhao, S. Zhang and B. Ye, J. Solid State Electrochem., 2011, 16, 505–511 CrossRef.
- M. Ghanei-Motlagh, M. Fayazi and M. A. Taher, Sens. Actuators, B, 2014, 199, 133–141 CrossRef CAS.
- S. M. J. M. Kermani, M. Ghanei-Motlagh, R. Zhiani, M. A. Taher, M. Fayazi and I. Razavipanah, J. Mol. Liq., 2015, 206, 145–150 CrossRef CAS.
- M. G. Motlagh, M. A. Taher and A. Ahmadi, Electrochim. Acta, 2010, 55, 6724–6730 CrossRef CAS.
- S. Morante-Zarcero, D. Pérez-Quintanilla and I. Sierra, J. Solid State Electrochem., 2015, 19, 2117–2127 CrossRef CAS.
- M. B. Gholivand, B. Geravandi and M. H. Parvin, Electroanalysis, 2011, 23, 1345–1351 CrossRef CAS.
- M. Fayazi, M. A. Taher, D. Afzali and A. Mostafavi, Sens. Actuators, B, 2016, 228, 1–9 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Corthey, E. Pensa, E. Cortes, M. H. Fonticelli, F. Ibanez, G. E. Benitez, P. Carro and R. C. Salvarezza, RSC Adv., 2014, 4, 27730–27754 RSC.
- C. S. Park, R. Colorado Jr, A. C. Jamison and T. R. Lee, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2016, DOI:10.1016/b978-0-12-803581-8.03780-2.
- J. Wei, Z. Guo, X. Chen, D.-D. Han, X.-K. Wang and X.-J. Huang, Anal. Chem., 2015, 87, 1991–1998 CrossRef CAS PubMed.
- R. K. Shervedani and S. A. Mozaffari, Anal. Chem., 2006, 78, 4957–4963 CrossRef CAS PubMed.
- F. Lucarelli, G. Marrazza, A. P. Turner and M. Mascini, Biosens. Bioelectron., 2004, 19, 515–530 CrossRef CAS PubMed.
- R. Segura, J. Pizarro, K. Díaz, A. Placencio, F. Godoy, E. Pino and F. Recio, Sens. Actuators, B, 2015, 220, 263–269 CrossRef CAS.
- J. Orozco, C. Fernández-Sánchez and C. Jiménez-Jorquera, Environ. Sci. Technol., 2008, 42, 4877–4882 CrossRef CAS PubMed.
- J. Zhou, L. Du, L. Zou, Y. Zou, N. Hu and P. Wang, Sens. Actuators, B, 2014, 197, 220–227 CrossRef CAS.
- L. Agüí, C. Peña-Farfal, P. Yáñez-Sedeño and J. M. Pingarrón, Talanta, 2007, 74, 412–420 CrossRef PubMed.
- M. Ghanei-Motlagh, M. A. Taher, A. Heydari, R. Ghanei-Motlagh and V. K. Gupta, Mater. Sci. Eng., C, 2016, 63, 367–375 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- T. Kuila, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Nanoscale, 2013, 5, 52–71 RSC.
- O.-K. Park, M. G. Hahm, S. Lee, H.-I. Joh, S.-I. Na, R. Vajtai, J. H. Lee, B.-C. Ku and P. M. Ajayan, Nano Lett., 2012, 12, 1789–1793 CrossRef CAS PubMed.
- J. B. Raoof, R. Ojani, A. Alinezhad and S. Z. Rezaie, Monatsh. Chem., 2010, 141, 279–284 CrossRef CAS.
- R. Y. A. Hassan, M. S. Kamel, H. N. A. Hassan and E. Khaled, J. Electroanal. Chem., 2015, 759, 101–106 CrossRef CAS.
- N. S. Gadhari, B. J. Sanghavi and A. K. Srivastava, Anal. Chim. Acta, 2011, 703, 31–40 CrossRef CAS PubMed.
- M. H. Mashhadizadeh, H. Khani, A. Shockravi and M. Sadeghpour, Mater. Sci. Eng., C, 2011, 31, 1674–1680 CrossRef CAS.
- N. S. Gadhari, B. J. Sanghavi, S. P. Karna and A. K. Srivastava, Electrochim. Acta, 2010, 56, 627–635 CrossRef CAS.
- R. Mohammadzadeh Kakhki and M. Rakhshanipour, Arabian J. Chem. DOI:10.1016/j.arabjc.2015.07.012.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- A. Alizadeh, M. M. Khodaei, C. Karami, M. S. Workentin, M. Shamsipur and M. Sadeghi, Nanotechnology, 2010, 21, 315503 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802, 10.1039/c39940000801.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- S. Srivastava, B. L. Frankamp and V. M. Rotello, Chem. Mater., 2005, 17, 487–490 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- A. Alizadeh, M. M. Khodaei, Z. Hamidi and M. b. Shamsuddin, Sens. Actuators, B, 2014, 190, 782–791 CrossRef CAS.
- M. Nasiri-Majd, M. A. Taher and H. Fazelirad, Talanta, 2015, 144, 204–209 CrossRef CAS PubMed.
- D. J. Fitzgerald, Regul. Toxicol. Pharmacol., 1995, 21, 177–179 CrossRef CAS PubMed.
- B. C. Janegitz, L. H. Marcolino-Junior, S. P. Campana-Filho, R. C. Faria and O. Fatibello-Filho, Sens. Actuators, B, 2009, 142, 260–266 CrossRef CAS.
- M. Etienne, J. Bessiere and A. Walcarius, Sens. Actuators, B, 2001, 76, 531–538 CrossRef CAS.
- W. Yantasee, Y. Lin, G. E. Fryxell and B. J. Busche, Anal. Chim. Acta, 2004, 502, 207–212 CrossRef CAS.
- W. Zhihua, L. Xiaole, Y. Jianming, Q. Yaxin and L. Xiaoquan, Electrochim. Acta, 2011, 58, 750–756 CrossRef.
- A. M. Ashrafi and K. Vytřas, Electrochim. Acta, 2012, 73, 112–117 CrossRef CAS.
- R. M. Takeuchi, A. L. Santos, P. M. Padilha and N. R. Stradiotto, Talanta, 2007, 71, 771–777 CrossRef CAS PubMed.
- A. Mohadesi and M. A. Taher, Talanta, 2007, 72, 95–100 CrossRef CAS PubMed.
- S. Kilinc Alpat, U. Yuksel and H. Akcay, Electrochem. Commun., 2005, 7, 130–134 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10267k |
| This journal is © The Royal Society of Chemistry 2016 |