
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe rare cis-configured trisubstituted lactam products obtained by the Castagnoli–Cushman reaction in N,N-dimethylformamide†
Dmitry
Dar'in
,
Olga
Bakulina
,
Sofia
Nikolskaya
,
Ivan
Gluzdikov
and
Mikhail
Krasavin
*
Institute of Chemistry, St. Petersburg State University, Peterhof 198504, Russian Federation. E-mail: m.krasavin@spbu.ru; Fax: +7 812 428 6939; Tel: +7 931 3617872
First published on 12th May 2016
Abstract
Unlike its trans counterpart, the cis-configured scaffold derived from the Castagnoli–Cushman reaction (CCR) is scarce and has not been explored in bioactive compound design. We found that conducting this reaction in DMF (in contrast to conventional toluene or xylene) led to a significantly higher proportion of cis-configured lactams in the diastereomeric product mixture. This allowed us, for the first time, to obtain and thoroughly characterize both stereoisomers of a significant number of lead-like CCR lactams. Simple rules for 1H NMR-based stereochemical assignment have been devised and correlated with the single-crystal X-ray structures obtained for pure cis- and trans-configured lactams.
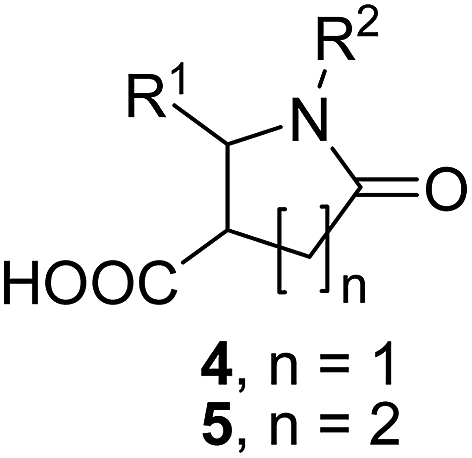
Discovered in 1969,1 the Castagnoli–Cushman reaction (CCR) of imines 1 (formed in situ or in a separate step), succinic (2) or glutaric (3) anhydrides provides convenient access to the medicinally important2 polysubstituted lactams 4–5 in a convenient, three-component format (Scheme 1).
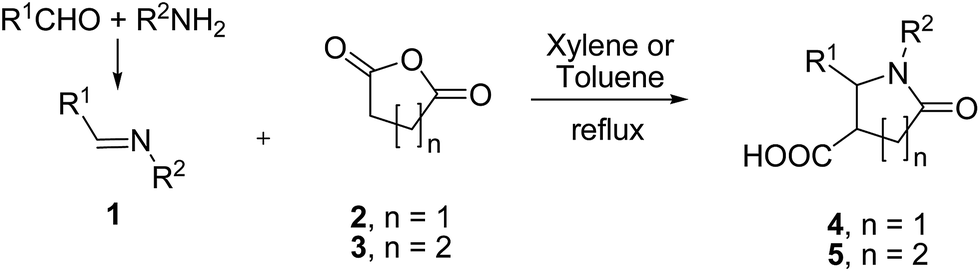 | ||
| Scheme 1 Castagnoli–Cushman reaction of succinic and glutaric anhydrides. | ||
CCR has recently emerged3 as an important synthetic tool for lead-oriented synthesis (LOS).4 Considering the challenges of inventing new LOS methods5 and our recent interest in developing lead-like compound libraries,6 particularly those employing the CCR,7 we thought it important to try and overcome certain limitations of the CCR, which were evident from the literature. Besides limitations of scope which we discuss elsewhere,8 several drawbacks associated with CCR are noteworthy. Firstly, in a vast majority of cases reported in the literature, the reaction involving simple anhydride inputs (such as 2–3) is conducted in low polarity aromatic hydrocarbon solvent (toluene or xylene) and requires rather forcing conditions (reflux). These two aspects introduce solubility and structural tolerability limitations, respectively.3 Secondly, almost universally the reaction was reported as proceeding with high diastereoselectivity (up to 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :52) and only the major, trans-isomer was isolated and characterized (with the exception of the pioneering reports by Castagnoli1 the minor, cis-isomer was isolated in <1% yield by fractional crystallization). Indirect synthesis of the cis counterpart of 4 (compound 6) via de-sulfurization (with reversal of configuration) of tetrasubstituted lactams 7 (obtainable, in turn, via four-component reaction of maleic anhydride) was reported by Shaw (Scheme 2).9 However, the cis-configured stereoisomer of 5 remain virtually unavailable. Multistep sequences leading to the cis-isomer of related δ-lactams have been reported by Stille10 and others.11 However, these strategies lack the practicality and flexibility offered by the multicomponent approach. From medicinal chemistry prospective, cis- and trans-versions of either 4 or 5 represent distinct scaffolds12 that ensure different spatial projection of the molecular periphery and, therefore, are likely to have vastly different complementarities to a protein target. The fact that the cis-isomer is always formed in impractical minority severely curbs the stereochemical diversity13 attainable by the CCR and represents a significant limitation of this chemistry.
:52) and only the major, trans-isomer was isolated and characterized (with the exception of the pioneering reports by Castagnoli1 the minor, cis-isomer was isolated in <1% yield by fractional crystallization). Indirect synthesis of the cis counterpart of 4 (compound 6) via de-sulfurization (with reversal of configuration) of tetrasubstituted lactams 7 (obtainable, in turn, via four-component reaction of maleic anhydride) was reported by Shaw (Scheme 2).9 However, the cis-configured stereoisomer of 5 remain virtually unavailable. Multistep sequences leading to the cis-isomer of related δ-lactams have been reported by Stille10 and others.11 However, these strategies lack the practicality and flexibility offered by the multicomponent approach. From medicinal chemistry prospective, cis- and trans-versions of either 4 or 5 represent distinct scaffolds12 that ensure different spatial projection of the molecular periphery and, therefore, are likely to have vastly different complementarities to a protein target. The fact that the cis-isomer is always formed in impractical minority severely curbs the stereochemical diversity13 attainable by the CCR and represents a significant limitation of this chemistry.
 | ||
| Scheme 2 Preparation of cis-configured γ-lactams reported by Shaw.9 | ||
In this work, we sought to find a solution to the above-mentioned limitations by drastically changing the solvent in which the CCR would be conducted. While toluene and xylene indeed appear to be solvents of choice (from the literature review), we ventured to investigate the CCR of more than two dozen three-component reactions in N,N-dimethylformamide (DMF).14 The choice of high-boiling DMF as a reaction medium in lieu of toluene or xylene was dictated by its higher polarity and ability to dissolve a wider range of reactants. In this Communication, we report the results of these studies.
A brief screening of various temperature regimens for the reactions leading to 4a and 5a (R1 = 4-MeOC6H4, R2 = Bn) revealed that changing the solvent medium from toluene to DMF offered little advantage in terms of lowering the reaction temperature. However, it was immediately evident that the formation of the cis-isomer was significantly more favored in DMF compared to toluene (Table S1 in ESI†). Particularly encouraging were the dr values obtained with glutaric anhydride at 110 °C (cis:trans = 1:2.5), considering the fact that, apart from the initial reports by Castagnoli and Cushman (vide supra),1cis-configured δ-lactams 5 were practically unattainable by the CCR. It should be noted that, occasionally, other solvents (such as DMSO) also favored the formation of the cis-configured products (see ESI†). However, this can more reliably achieved with DMF as a solvent of choice.
Encouraged by these results, we extended the new reaction protocol to a range of Schiff bases (pre-formed by reaction of a primary amine with an aldehyde in presence of MgSO4 or molecular sieves) which underwent CCR with succinic (1) and glutaric (2) anhydrides at 110 °C in DMF over 5–24 h period (Scheme 3).
 | ||
| Scheme 3 The new CCR protocol developed in this work. | ||
The reaction times shown were required for the reactions to proceed to completion. The dr values were determined by the relative integration of characteristic signals in the 1H NMR spectrum of the crude reaction mixture. The isolated yields were calculated for the analytically pure mixture of cis- and trans-4(5) in which the ratio of diastereomers remained practically unchanged compared to that in the crude reaction mixture (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | n | Isolated yield (%) | Reaction time | dr (cis:trans) |
| a Reaction temperature was raised to 125 °C in order to ensure maximum conversion. | ||||||
| 4a | 4-MeOC6H4 | Bn | 1 | 74 | 5 h | 1:3.5 |
| 4b | 4-MeOC6H4 | i-Pr | 1 | 75 | 5 h | 1:1.8 |
| 4c | 4-MeOC6H4 | t-Bu | 1 | 69 | 5 h | 1:1.7 |
| 4d | 4-FC6H4 | n-Pr | 1 | 46 | 5 h | 1:2.5 |
| 4e | 4-MeC6H4 | Et | 1 | 70 | 5 h | 1:2 |
| 4f | 4-BrC6H4 | Bn | 1 | 48 | 24 h | 1:3 |
| 4g | 4-O2NC6H4 | n-Bu | 1 | 28 | 24 h | 1:4.5 |
| 4h | 3-Py | n-Pr | 1 | 62 | 6 h | 1:2 |
| 4i | 3-O2NC6H4 | Me | 1 | 48 | 6 h | 1:1.8 |
| 4j | 4-Me2NC6H4 | Allyl | 1 | 84 | 6 h | 1:2 |
| 4k | 1-Naphthyl | n-Pr | 1 | 59 | 6 h | 1:2.1 |
| 5a | 4-MeOC6H4 | Bn | 2 | 66 | 24 h | 1:2 |
| 5b | 4-MeOC6H4 | i-Pr | 2 | 64 | 6 h | 1:1.8 |
| 5c | 4-MeOC6H4 | t-Bu | 2 | 59 | 6 h | 1:1.4 |
| 5d | 4-FC6H4 | n-Pr | 2 | 51 | 6 h | 1:2 |
| 5e | 2-Thienyl | cyclo-Pr | 2 | 35 | 7 days | 1:2.3 |
| 5f | 4-MeC6H4 | Et | 2 | 53 | 6 h | 1:2.2 |
| 5g | 3,4-(MeO)2C6H3 | n-Bu | 2 | 62 | 6 h | 1:2 |
| 5h | 4-O2NC6H4 | n-Bu | 2 | 16 | 24 h | 1:2.2 |
| 5i | 2-MeOC6H4 | Bn | 2 | 52 | 24 h | 1:1.7 |
| 5j | 1-Naphthyl | n-Pr | 2 | 58 | 24 h | 1:3.6 |
| 5k | 4-AcNHC6H4 | cyclo-Pr | 2 | 54 | 48 h | 1:2.3 |
| 5l | 3-Py | n-Pr | 2 | 61 | 24 h | 1:2.5 |
| 5m | Ph | Ph | 2 | 39 | 22 ha | 1:5 |
| 5n | 4-MeC6H4 | 4-MeOC6H4 | 2 | 47 | 22 ha | 1:4.2 |
| 5o | 4-MeOC6H4 | 4-MeOC6H4 | 2 | 53 | 22 ha | 1:4.1 |
Some general observations can be made based on the results of the substrate survey. The outcome of the reaction seems to be independent of the steric bulk of around the imine nitrogen (R2), which gives rise to the lactam nitrogen in the reaction product (cf. isolated yields of 5c and 5d, 4a and 4c). However, the reaction appears to be rather sensitive to the substituent effects – with electron-deficient groups drastically reducing the product yields (4g and 5h), which is consistent with the earlier observations by Cushman.15 Imines derived from anilines (such as those used in preparation of 5m–o) were expectedly less reactive compared to their counterparts carrying an aliphatic amine portion (which may explain the scarcity of the respective CCR products reported in the literature).16 For these bis-aryl substrates, the reaction temperature was raised to 125 °C and the reaction time extended to 22–24 h, in order to reach maximum conversion. This, in turn, was associated with lower proportion of the cis-configured δ-lactam in the product mixture.
Considering the latter observation, we were curious to see if prolonged reaction times can could cause a ‘drift’ in the observed dr values, via a gradual isomerization of the cis-isomer into the thermodynamically more stable trans-isomer (a transformation certainly achievable with good yield on treatment with a strong base, such as potassium tert-butoxide).9 To verify that, diastereomeric mixtures of 4a (cis/trans = 1:3.5) and 5a (cis/trans = 1:2) were heated in DMF at 110 °C for a period of up to 10 days with periodic monitoring of the dr. As the result, the dr of 4a changed drastically to 1:13.8 while 5a was somewhat more resistant to thermal isomerization: its dr only changed to 1:3.9. This, however, clearly indicates that, in order to maximize the formation of the cis-isomer, the CCR should be closely monitored and the reactions should be stopped as soon as the maximum conversion is reached (the data reported in Table 1 was obtained in this manner).
Formation of a higher proportion of the cis-isomer of lactams 4 and 5 allowed, for the first time, separation, on preparative scale, and full characterization of cis- and trans-isomers of compounds presented in Table 1 (see ESI†). There has not been much progress in comparative characterization of diastereomeric CCR products since the pioneering work of Castagnoli and Cushman.1 Therefore, in this work we also aimed to establish, using the wide range of compounds synthesized, a basis for assigning cis- or trans-configuration to the γ- and δ-lactams obtainable by the CCR based on their NMR spectra and to correlate these findings with a single-crystal X-ray crystallography data.
As it is evident for the value of the coupling constants of the C2–H protons provide a rather solid basis for stereochemical assignment of γ-lactams 4a–j, as was also reported earlier.1,3 However, the difference in the same coupling constants is not that pronounced for δ-lactams 5a–o and varies from 0.5 to 2.0 Hz (though the value appears to be always larger for the cis-isomer compared to its trans-counterpart). In an attempt to look for more characteristic patterns that could additionally aid in the stereochemical assignment, we also looked at the chemical shifts of C2–H and C3–H protons. While the former displayed no characteristic pattern, the latter appear to be universally positioned downfield for the cis-isomer compared to the trans-isomer. This difference also appears to be more pronounced for of γ-lactams 4 compared to δ-lactams 5 (Table 2). In order to ultimately confirm our stereochemical assignment (which was initially based on the dr values observed and then confirmed by the 1H NMR patterns displayed by the respective stereoisomers, as discussed above), we obtained a number of single-crystal X-ray structures of individual cis- and trans-isomers of the CCR lactams in both 4 and 5 series (see ESI,† a representative pair is shown in Fig. 1). To the best of our knowledge, until today, crystallographic information on the cis-isomers of the CCR lactams (whilom scarcely available) has been lacking in the literature.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | 3 J (C2–H), Hz | δ (C2–H), ppm (multiplicity) | δ (C3–H), ppm (multiplicity) | Δ [δ(C3–H)] cis/trans, ppm | |||
| cis | trans | cis | trans | cis | trans | ||
| a Stereochemical assignment confirmed by single-crystal X-ray analysis. | |||||||
| 4a | 9.0 | 5.8 | 4.64 (d) | 4.50 (d) | 3.54 (dt) | 3.06 (ddd) | 0.48 |
| 4b | 9.1a | 4.4a | 4.97 (d) | 4.79 (d) | 3.61 (dt) | 2.89 (ddd) | 0.72 |
| 4c | 8.6a | 1.0 | 5.13 (d) | 5.09 (d) | 3.53 (dt) | 2.62 (ddd) | 0.91 |
| 4d | 9.2a | 5.6a | 5.00 (d) | 4.81 (d) | 3.66 (dt) | 3.02 (ddd) | 0.64 |
| 4e | 9.3a | 5.9 | 4.99 (d) | 4.76 (d) | 3.63 (dt) | 3.00 (ddd) | 0.63 |
| 4f | 9.3 | 5.5 | 4.72 (d) | 4.55 (d) | 3.66 (dt) | 3.07 (ddd) | 0.59 |
| 4g | 9.2 | 5.4 | 5.17 (d) | 4.99 (d) | 3.72 (dt) | 3.06 (ddd) | 0.66 |
| 4h | 9.3 | 5.7 | 5.06 (d) | 4.85 (d) | 3.73 (dt) | 3.11 (ddd) | 0.62 |
| 4i | 9.3 | 6.2 | 5.13 (d) | 4.94 (d) | 3.71 (dt) | 3.11 (ddd) | 0.60 |
| 4j | 9.0 | 5.7 | 4.77 (d) | 4.62 (d) | 3.64 (dt) | 3.01 (ddd) | 0.63 |
| 5a | 5.0 | 5.6 | 4.71 (d) | 4.70 (d) | 3.11 (ddd) | 2.78 (m) | 0.33 |
| 5b | 4.6a | 2.8 | 4.93 (d) | 4.98 (d) | 3.00 (dt) | 2.72 (m) | 0.29 |
| 5c | 4.1a | 2.1 | 5.22 (d) | 5.38 (d) | 3.01 (dt) | 2.79 (dt) | 0.30 |
| 5d | 5.1 | 4.1 | 4.95 (d) | 4.97 (d) | 3.18 (m) | 2.82 (dt) | 0.36 |
| 5e | 4.7 | 3.6 | 5.17 (d) | 5.19 (d) | 3.25 (m) | 2.95 (m) | 0.18 |
| 5f | 4.5a | 4.2 | 4.91 (d) | 4.90 (d) | 3.12 (m) | 2.79 (dt) | 0.33 |
| 5g | 4.6 | 4.1 | 4.88 (d) | 4.89 (d) | 3.10 (m) | 2.85 (dt) | 0.25 |
| 5h | 5.2 | 3.7 | 5.18 (d) | 5.15 (d) | 3.16 (m) | 2.88 (m) | 0.27 |
| 5i | 4.9 | 4.0 | 5.10 (d) | 5.17 (d) | 3.04 (ddd) | 2.81 (dt) | 0.23 |
| 5j | 5.4 | 2.3 | 5.91 (d) | 5.85 (br s) | 3.33 (m) | 2.98 (dt) | 0.35 |
| 5k | 4.9 | 4.3 | 4.84 (d) | 4.87 (d) | 3.08 (m) | 2.78 (dt) | 0.30 |
| 5l | 5.2 | 4.2 | 5.20 (d) | 5.04 (d) | 3.26 (ddd) | 2.89 (dt) | 0.37 |
| 5m | 5.0 | 4.2 | 5.29 (d) | 5.39 (d) | 3.49 (ddd) | 2.98 (dt) | 0.51 |
| 5n | 4.9a | 4.2 | 5.17 (d) | 5.26 (d) | 3.45 (ddd) | 2.91 (dt) | 0.54 |
| 5o | 5.1 | 4.5 | 5.16 (d) | 5.23 (d) | 3.44 (ddd) | 2.91 (dt) | 0.53 |

 | ||
| Fig. 1 Single-crystal X-ray structures of cis- and trans-isomers (left and right, respectively) of compound 4d. | ||
The individual cis- and trans-isomers reported in this Communication were obtained by preparative reverse-phase HPLC separation of diastereomeric mixtures of carboxylic acid lactams 4 and 5 (which are difficult to separate by conventional column chromatography). Alternatively, the carboxylic acids can be converted to the respective methyl esters, in order to facilitate chromatographic separation on silica gel.9 This strategy was successfully realized for compound 5d (Scheme 4).
 | ||
| Scheme 4 Diastereomer separation of methyl esters obtained from compound 5d. | ||
In conclusion, we established that the Castagnoli–Cushman reaction in DMF leads to higher yields of the cis-configured γ- and δ-lactams. This enabled preparative isolation and characterization of this rare type of lead-like scaffolds in comparison with their well-described trans-configured counterparts. This, in turn, led to a more reliable basis for stereochemical assignment based on characteristic 1H NMR patterns displayed by the two isomeric series, which was correlated with a number of X-ray structures. Efforts to unveil the medicinal chemistry potential of the cis-isomeric CCR lactams are underway in our laboratories. The results will be reported in due course.
Acknowledgements
We gratefully acknowledge support from the Russian Scientific Fund (Project Grant 14-50-00069). NMR studies, X-ray analysis and Mass Spectrometry studies were performed at the Research Center for Magnetic Resonance, the Research Center for X-ray Diffraction Studies and the Center for Chemical Analysis and Materials Research of Saint-Petersburg State University.Notes and references
- (a) N. Castagnoli, J. Org. Chem., 1969, 34, 3187–3189 CrossRef CAS PubMed; (b) M. Cushman and N. Castagnoli, J. Org. Chem., 1973, 38, 440–448 CrossRef CAS PubMed; (c) M. Cushman and N. Castagnoli, J. Org. Chem., 1974, 39, 1546–1550 CrossRef CAS PubMed.
- M. González-López and J. T. Shaw, Chem. Rev., 2009, 108, 164–189 CrossRef PubMed.
- S. V. Ryabukhin, D. M. Panov, D. S. Granat, E. N. Ostapchuk, D. V. Kryvoruchko and O. O. Grygorenko, ACS Comb. Sci., 2014, 16, 146–153 CrossRef CAS PubMed.
- A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114–1122 CrossRef CAS PubMed.
- R. Doveston, S. Marsden and A. Nelson, Drug Discovery Today, 2014, 19, 813–819 CrossRef CAS PubMed.
- (a) A. Mishchuk, N. Shtil, M. Poberezhnyk, K. Nazarenko, T. Savchenko, A. Tolmachev and M. Krasavin, Tetrahedron Lett., 2016, 57, 1056–1059 CrossRef CAS; (b) P. Mujumdar, P. Sarnpitak, A. Shetnev, M. Dorogov and M. Krasavin, Tetrahedron Lett., 2015, 56, 2827–2831 CrossRef CAS.
- D. Dar'in, O. Bakulina, M. Chizhova and M. Krasavin, Org. Lett., 2015, 17, 3930–3933 CrossRef PubMed.
- M. Krasavin and D. Dar'in, Tetrahedron Lett., 2016, 57, 1635–1640 CrossRef CAS.
- J. Wei and J. T. Shaw, Org. Lett., 2007, 9, 4077–4080 CrossRef CAS PubMed.
- (a) K. Paulvannan, J. B. Schwarz and J. R. Stille, Tetrahedron Lett., 1993, 34, 215–218 CrossRef CAS; (b) K. Paulvannan and J. R. Stille, Tetrahedron Lett., 1993, 34, 8197–8200 CrossRef CAS; (c) K. Paulvannan and J. R. Stille, J. Org. Chem., 1994, 59, 1613–1620 CrossRef CAS; (d) G. R. Cook, L. G. Beholz and J. R. Stille, J. Org. Chem., 1994, 59, 3575–3584 CrossRef CAS.
- (a) A. Samarat, J. B. Kraïem, T. Ben Ayed and H. Amri, Tetrahedron, 2008, 64, 9540–9543 CrossRef CAS; (b) N. M. Garrido, M. R. Sanchez, D. Diez, F. Sanz and J. G. Urones, Tetrahedron: Asymmetry, 2011, 22, 872–880 CrossRef CAS.
- C. Agami, L. Hamon, C. Kadouri-Puchot and V. Le Guen, J. Org. Chem., 1996, 61, 5736–5742 CrossRef CAS.
- P. Y. Ng, Y. Tang, W. M. Knosp, H. S. Stadler and J. T. Shaw, Angew. Chem., Int. Ed., 2007, 46, 5352–5355 CrossRef CAS PubMed.
- The CCR of homophthalic anhydride (but not of 2 or 3) was investigated in different solvents (CHCl3, MeOH and HCONH2): M. Cushman and E. J. Madaj, J. Org. Chem., 1987, 52, 907–915 CrossRef CAS.
- M. Cushman and E. J. Madaj, J. Org. Chem., 1987, 52, 907–915 CrossRef CAS.
- According to SciFinder search performed in February 2016, only six examples of bis-aryl piperidones similar to 5m–o have been reported: (a) T. Mani, D. Liu, D. Zhou, L. Li, W. E. Knabe, F. Wang, K. Oh and S. O. Meroueh, ChemMedChem, 2013, 8, 1963–1977 CrossRef CAS PubMed; (b) M. Tabcheh, M. Baroudi, F. Elomar, A. Elzant, M. Elkhatib and V. Rolland, Asian J. Chem., 2006, 18, 1771–1782 CAS; (c) J. Robert, A. Boucherle and C. Luu-Duc, J. Pharm. Belg., 1989, 44, 36–40 CAS. The same search identified less than one hundred γ-lactams related to 4, many of which had been prepared using methods other than CCR: (d) Z. Li, Y. Feng, Z. Li and J. Lan, Synlett, 2014, 25, 2899–2902 CrossRef CAS; (e) M. Pohmakotr, N. Yotapan, P. Tuchinda, C. Kuhakarn and V. Reutrakul, J. Org. Chem., 2007, 72, 5016–5019 CrossRef CAS PubMed; (f) M. Pohmakotr, N. Yotapan, P. Tuchinda, C. Kuhakarn and V. Reutrakul, Tetrahedron, 2007, 63, 4328–4337 CrossRef CAS; (g) X. Zhao, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 12466–12469 CrossRef CAS PubMed; (h) M. Tang, D. Xing, H. Huang and W. Hu, Chem. Commun., 2015, 51, 10612–10615 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1438073, 1442379, 1442947, 1447518, 1447521, 1437633, 1442993, 1442357, 1442376 and 1452491. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra10249b |
| This journal is © The Royal Society of Chemistry 2016 |