
A fully automated portable gas chromatography system for sensitive and rapid quantification of volatile organic compounds in water†
Abstract
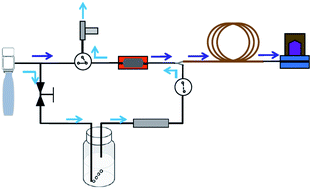
This paper presents the design, assembly and characterization of a fully automated portable gas chromatography system coupled with a purge-and-trap system for the sensitive and rapid field analysis of volatile organic compounds (VOCs) in water samples. The VOCs were firstly purged by helium gas in the micro-fabricated preconcentrator/injector and then injected into the downstream capillary column and photoionization detector for separation and detection. The purge-and-trap conditions were optimized to efficiently extract VOCs from water samples. The calibration of 6 VOCs with concentrations ranging from 1 μg L−1 to 500 μg L−1 showed excellent linearity (R2 > 0.99). Detection limits (3σ) of sub-μg L−1 (or sub-parts-per-billion level) were achieved, which are orders of magnitude lower than the maximum contaminant level (MCL) established by the US Environmental Protection Agency (EPA). The separation of 26 analytes (in a vapor pressure range from 0.087 Torr to 180 Torr) in a water sample in less than 15 minutes was also demonstrated. Finally, the optimized system was applied to field analysis of a groundwater sample in an environmental remediation site. The quantified results agreed well with those obtained by an analytical lab using standard analytical methods and instruments. Our system offers a lab-on-a-chip solution for sensitive and rapid water analysis compliant with the EPA sample collection method. It will have a wide range of applications in environmental monitoring, industries and healthcare.
 Please wait while we load your content...
Please wait while we load your content...

