
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanomechanical properties of protein–DNA layers with different oligonucleotide tethers†
Cristina Gutiérrez
Sánchez‡
,
Qiang
Su‡
,
Sabine
Wenderhold-Reeb
and
Gilbert
Nöll
*
Nöll Junior Research Group, Organic Chemistry, Chem. Biol. Dept., Faculty IV, Siegen University, Adolf-Reichwein-Str. 2, 57068 Siegen, Germany. E-mail: noell@chemie.uni-siegen.de
First published on 6th June 2016
Abstract
The multi-ligand binding flavoprotein dodecin is reconstituted on top of flavin-terminated oligonucleotide monolayers. A detailed quartz crystal microbalance with a dissipation monitoring (QCM-D) study showing how the length and flexibility of the oligonucleotide tethers influence the stability and the viscoelastic properties of the resulting DNA–protein layers is presented. Relatively dense protein layers can be obtained, if the length of the tethers is in the same range as the diameter of dodecin. When significantly longer tethers are used, less dense layers are formed. When rather short tethers are used, the reaching area of individual tethers is too low to capture single apododecin molecules cooperatively, and the formation of stable and dense protein layers is not possible. On top of the DNA–dodecin layers additional flavin–DNA ligands may be captured to form sandwich-type DNA–protein–DNA layers. Differences in the binding and unbinding behavior of flavin-dsDNA and flavin-ssDNA ligands are measured by QCM-D and surface plasmon fluorescence spectroscopy (SPFS). Both type of ligands show relatively low kon values, which might be explained by the structural rigidity of the binding pockets allowing a ligand to enter only when it approaches precisely in the right orientation. Apparently apododecin–flavin binding follows Fischer's classic lock-and-key binding model.
Introduction
Recently the multi-ligand binding flavoprotein dodecin from Halobacterium salinarum has been introduced as a key element for reversible surface modification in nano-biotechnology allowing the multiple generation, erasure and reprogramming of sandwich-type DNA–protein–DNA nanostructures on surfaces.1 Dodecin is a dodecameric hollow-spherical riboflavin-binding protein with a diameter of about 7 nm that comprises six binding pockets with an octahedral arrangement.2–9 Since each binding pocket can bind up to two flavin ligands, dodecin is able to bind up to twelve flavins in total. For a single apododecin–flavin bond a dissociation constant of Kd = 580 nM was calculated from the kinetic binding and unbinding constants of kon (QCM) = 7.7 × 103 M−1 s−1 and koff (QCM) = 4.5 × 10−3 s−1 determined by quartz crystal microbalance with dissipation (QCM-D) measurements. For these measurements the adsorption and desorption of the multivalent tE variant of apododecin (DtE) at a flavin-terminated DNA–monolayer with rather low flavin surface coverage was investigated. To minimize the probability for multi-ligand binding, first a layer of (dithiane)3-anchored single stranded DNA (ssDNA) of 20 bases was formed on gold and subsequently treated with a mixture of short thiols (to remove non-specifically adsorbed ssDNA), before it was hybridized with a mixture of flavin-modified and flavin-free complementary DNA at a ratio of 1 to 9 leading to a relative flavin surface coverage of 10% with respect to the total amount of double-stranded DNA (dsDNA).1 With respect to the half-life of τ ≈ 3 min calculated from koff, a single apododecin–flavin bond is not sufficiently stable for long-term surface modification. Nevertheless stable dodecin layers could be formed via multi-ligand binding at a relative flavin surface coverage of 100%. These protein layers were further modified by adsorption of bidentate flavin ligands resulting in stable sandwich-type DNA–protein–DNA nanostructures. Since dodecin binds flavin ligands with high affinity when they are oxidized, whereas flavin reduction induces the dissociation of the holoprotein in apododecin and free flavin ligands,2,5,7 the generated nanostructures could be disassembled by chemical reduction using a buffered solution of sodium dithionite and reprogrammed by adsorption of a different type of bidentate flavin anchored ligand after reassembly of the dodecin layer.1 Based on this previous study apododecin–flavin could evolve into an interesting alternative to streptavidin–biotin, which is probably the most widely applied multi-ligand binding system for surface modification. Using streptavidin–biotin the impact of tether length and dynamics in modulating receptor–ligand binding has been investigated.10–13 It turned out that not only the ligand–receptor pair interaction but also the length and the flexibility of the tethering chains determine the overall range, rate, and ultimate strength of complementary multiple bond formation. Similar observations were made when the interaction between DNA coated emulsion droplets equipped with complementary sticky ends was studied.14While in previous studies regarding the reconstitution of dodecin on surfaces always dsDNA tethers comprising 20 base pairs (bp) were used,1,2,5 here a detailed QCM-D study is presented showing how the length and flexibility of the surface grafted and flavin-terminated oligonucleotide tethers influence the stability and the viscoelastic properties of the resulting DNA–protein layers, which may then be used to bind additional bi- or multidentate ligands. In addition differences in the binding and unbinding behavior of flavin-dsDNA and flavin-ssDNA ligands are measured by QCM-D and surface plasmon fluorescence spectroscopy (SPFS). Both type of ligands show relatively low kon values for apdodecin–flavin complex formation in comparison to the streptavidin–biotin system, which follows almost diffusion-controlled kinetics. The different kinetics might be explained by differences in the ligand binding mechanism of both proteins. The apododecin–flavin ligand binding kinetics are discussed in the context of the lock and key model, which has been introduced in 1894 by Emil Fischer.15
Experimental section
Chemicals
Chemicals for buffer preparation (NaCl, Tris, MgCl2) were purchased from Roth Chemicals. In our experiments we used a buffer solution at pH = 7.5, containing 1 M of NaCl, 20 mM of Tris–HCl, and 5 mM of MgCl2. Mercaptobutanol (MCB) and mercaptopropionic acid (MPA) were purchased from Sigma-Aldrich. All oligomers were purchased from Eurogentec S.A. Liège Science Park, (Seraing Belgium) or Metabion GmbH, (Martinsried, Germany). For QCM-D and SPFS measurements ssDNA comprising 20 bases (5′-AAC-TAC-TGG-GCC-ATC-GTG-AC-3′) modified at the 5′-end with three dithiane groups introduced by the use of dithiol-phosphoramidite (6750.6 g mol−1), as well as complementary ssDNA modified with a flavin (CofC4) at the 5′-end (6472.4 g mol−1) were used.1,5 Moreover, ssDNA (5′-GTC-ACG-ATG-GCC-CAG-TAG-TT-3′) modified with a flavin (CofC4) at the 5′ end and one dithiane ring at the 3′ end was purchased (6672.4 g mol−1). For SPR, SPFS, and QCM-D measurements ssDNA (5′-AAC-TAC-TGG-GCC-ATC-GTG-AC-3′) labeled with the fluorescent dye Cy5® at the 5′ end was hybridized with complementary flavin-modified ssDNA mentioned above to form a monodentate Cy5® labeled flavin-dsDNA ligand. Furthermore a flavin-Cy5® modified ssDNA oligomer (5′-GTC-ACG-ATG-GCC-CAG-TAG-TT-3′) with the flavin (CofC6) at the 5′ end and Cy5® at the 3′ end was purchased (7347.6 g mol−1) and used as monodentate Cy5® labeled flavin-ssDNA ligand. To elongate the dsDNA tethers from 20 to 100 bp a ssDNA fragment with 80 bases (5′-CGC-CGC-CGC-CGA-CGA-CGC-CAT-CGA-CCG-CGC-GGA-GGA-CAC-CCT-CGA-CAA-CGT-GGT-GTG-GGC-GTC-ACG-ATG-GCC-CAG-TAG-TT-3′) (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 639.9 g mol−1), was hybridized with a partially (60 bases/80 bases) complementary ssDNA (5′-GCC-CAC-ACC-ACG-TTG-TCG-AGG-GTG-TCC-TCC-GCG-CGG-TCG-ATG-GCG-TCG-TCG-GCG-GCG-GCG-AAC-TAC-TGG-GCC-ATC-GTG-AC-3′) (24683.9 g mol−1). Besides modified DNA, dsLNA was applied as a tether. For this purpose ssLNA with 7 bases (5′-GTC-GCC-C-3′) was modified with 1,2-dithiane at the 5′ end (2525.7 g mol−1) and complementary ssLNA (5′-GGG-CGA-C-3′) with CofC4-flavin at the 5′ end (2709.9 g mol−1), respectively.
639.9 g mol−1), was hybridized with a partially (60 bases/80 bases) complementary ssDNA (5′-GCC-CAC-ACC-ACG-TTG-TCG-AGG-GTG-TCC-TCC-GCG-CGG-TCG-ATG-GCG-TCG-TCG-GCG-GCG-GCG-AAC-TAC-TGG-GCC-ATC-GTG-AC-3′) (24683.9 g mol−1). Besides modified DNA, dsLNA was applied as a tether. For this purpose ssLNA with 7 bases (5′-GTC-GCC-C-3′) was modified with 1,2-dithiane at the 5′ end (2525.7 g mol−1) and complementary ssLNA (5′-GGG-CGA-C-3′) with CofC4-flavin at the 5′ end (2709.9 g mol−1), respectively.
Expression, purification and refolding of heterologously expressed apododecin variants
The heterologous overexpression of N-terminally His6 tagged halopilic apododecin flavin binding variant triple E (tE; E45A delE50 delE51) and a nonbinding variant W36 (W36AQ55A) was performed by transformation of the recombinant plasmid pDOD (pET22b(+), Novagen) with subcloned tE PCR product into chemocompetent BL21 (De3) E. coli strain. Purification and refolding processes were done as described previously.3 Dodecameric dodecin was detected by size exclusion chromatography on a HiLoad 16/600 Superdex 200 pg column (GE Healthcare Life Sciences) equilibrated and eluted with buffer.Preparation of the gold substrates
:1 concentrated H2SO4/30%H2O2, CAUTION: piranha solution reacts violently with most organic materials and must be handled with extreme care) for 5 min at room temperature and then rinsed with pure water.
For the QCM-D measurements standard sensor chips (QSX301, Q-Sense, Västra Frölunda, Sweden) with the following specifications were used: frequency 4.95 MHz ± 50 kHz, diameter 14 mm, thickness 0.3 mm, RMS surface roughness of electrode <3 nm. Before modification, the sensors were cleaned with an UV cleaner for 10 min, thereafter with basic Piranha (1:1:5 of H2O2, 25% ammonia solution, pure water) at 75 °C for 5 min, and again with UV treatment for 10 min.
Deposition of DNA, apododecin, and ligands
The thiolated ssDNA was adsorbed on the SPR and QCM-D chips in the following way. The DNA was dissolved in buffer and adsorbed to the gold surface for 1 h. The 1,2-dithiane and flavin modified ssDNA was used at a concentration of 2 μM, and the (1,2-dithiane)3 modified ssDNA at 5 μM, respectively. Efficient chemisorption of the DNA strands to the gold surface was accomplished via the reaction of one or three dithiane groups with gold. After adsorption was completed, the surface was rinsed with buffer and pure water. To replace non-specifically adsorbed DNA strands and saturate free sites at the gold surface, in the next step a mixture of low-molecular weight thiols was adsorbed.16 Equimolar amounts of mercaptobutanol (MCB) and mercaptopropionic acid (MPA), dissolved in water at 2 mM overall concentration, were adsorbed on the DNA–gold surface for 30 min followed by rinsing with pure water. In order to obtain the flavin-dsDNA and dsLNA ligand modified surface, the surface grafted ssDNA and ssLNA was hybridized with complementary flavin-modified ssDNA and ssLNA at 5 μM in buffer. In the process of 100 bp DNA-tether fabrication, the (dithiane)3-modified ssDNA (5 μM) was adsorbed on gold (followed by adsorption of a mixture of MCB and MPA, step 1), then subsequently hybridized with two oligomers of 80 bases (1 μM each, steps 2 and 3), and thereafter with flavin modified ssDNA (3.2 μM, step 4). A cartoon visualizing this approach is depicted in Scheme 1. In all experiments apododecin DtE was added at a concentration of 5 μM. The monodentate flavin ligands were added at a concentration of 2.5 μM. Each step of surface modification was monitored by SPR or QCM-D measurements. | ||
| Scheme 1 Strategy for the stepwise assembly of a flavin dsDNA (100 bp) ligand. | ||
SPR, SPFS and QCM-D
For SPR and SPFS a commercially available setup from Res-Tec (Resonant Technologies, Framersheim, Germany) was used. QCM-D measurements were performed with a Q-sense E1 instrument (Q-Sense, Göteborg, Sweden) which was controlled by the QSoft 401 acquisition software. The QCM sensor crystals were cleaned in a BioForce ozone cleaner (BioForce, Ames, USA). Unless otherwise stated, rinsing with buffer was done at a flow rate of 250 μL min−1.Results and discussion
Effect of tether flexibility
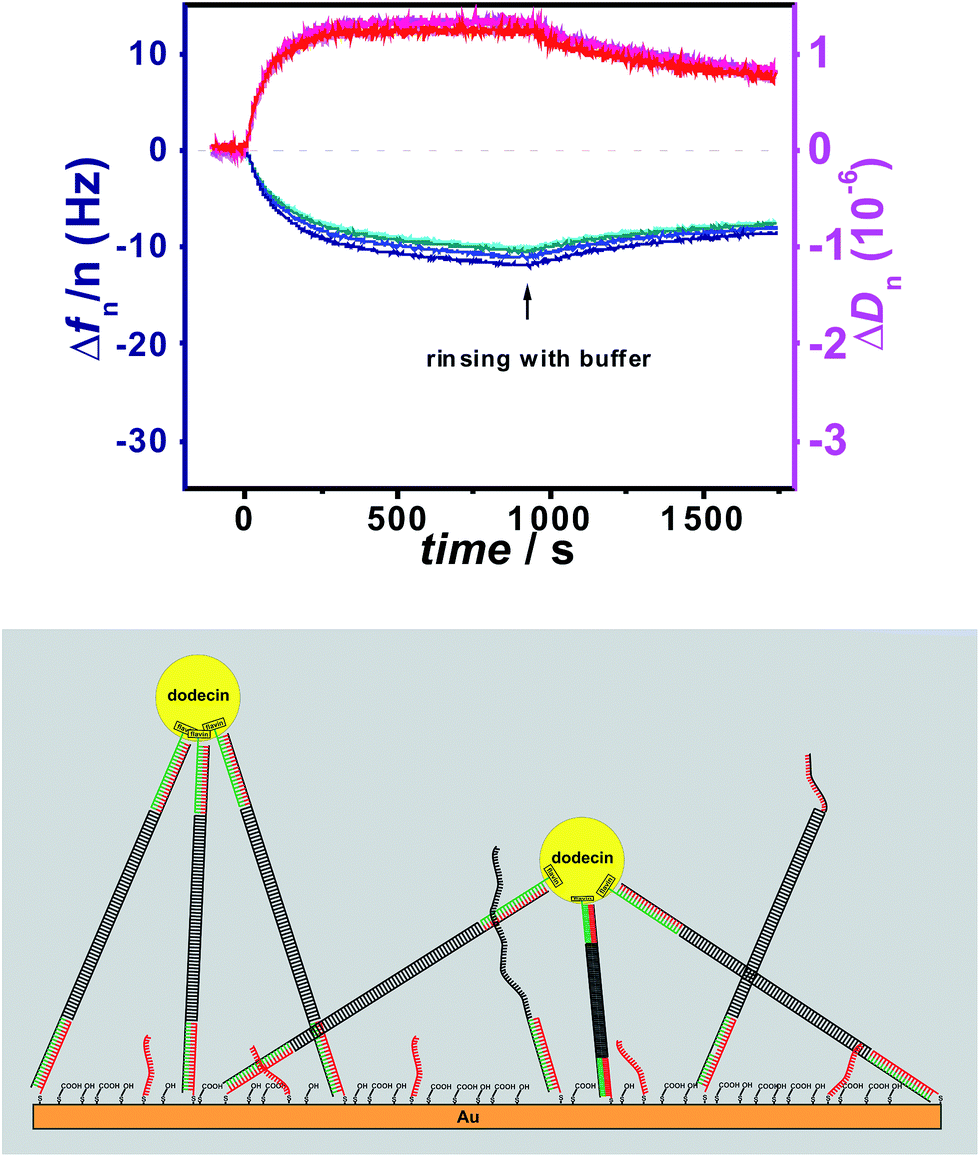
To study the effect of tether flexibility a 20 bp comprising dsDNA tether used previously,1 is compared with a more flexible tether of ssDNA with 20 bases. A tether length of 20 bp was chosen, since the length of dsDNA with 20 bp is almost 7 nm, which is in the same range as the diameter of dodecin. While dsDNA of 20 bp behaves like a rigid rod (for dsDNA the mechanical persistence length is 50 nm),17–19 ssDNA is highly flexible (with a persistence length of less than 2 nm).20 Using both tethers, flavin-terminated DNA layers were formed at which apododecin DtE was then adsorbed, and the protein layer was subsequently rinsed with buffer solution. Further details are given in the experimental section. The corresponding QCM-D binding and unbinding curves are shown in Fig. 1 and S1 in ESI† using a different presentation in which the bandwidth (Γ) instead of the dissipation factor (D) is shown. | ||
| Fig. 1 Frequency (blue) and dissipation factor (red) shifts (overtones 5-13) observed in situ by QCM-D for adsorption/desorption of apododecin DtE on monolayers of flavin-terminated dsDNA with 20 bp (A) and ssDNA with 20 bases (B) as shown in the cartoon below. | ||
On the surface modified with flavin-dsDNA ligands dodecin forms a rigid layer with a typical overlap of the frequency overtones (see Fig. 1A and 2B in ref. 1). Initially there is an increase of the dissipation signal caused by rocking and sliding motions of apododecin adsorbed mainly by single flavin ligands before the dissipation factor decreases with proceeding formation of multi-ligand binding leading to a stiff layer with restricted lateral motion of the adsorbed apoprotein molecules.1 The fact that dissipation-factor values for lower overtones decrease below the initial level prior to apododecin adsorption indicates that the lateral motion of the dsDNA tethers is also being restricted upon binding with apododecin.21–24 Considerable spreading of the dissipation overtones indicates strong viscoelastic dispersion, which may originate from relaxation processes at rates comparable to the oscillation frequency of the resonator.23,25
Adsorption of apododecin on the surface modified with flavin-ssDNA tethers also results in the formation of a rigid layer (see Fig. 1B and S1B†). The somewhat lower shift in frequency in comparison to the dsDNA tethers can be explained by a lower amount of water molecules and possibly also cations being trapped in the ssDNA sublayer than in the dsDNA sublayer.23 In contrast to the flavin-dsDNA tethers, for the flavin-ssDNA tethers the bandwidth shift of the 5th through 13th overtones shows little difference during apododecin adsorption.
Effect of tether length
In order to evaluate the effect of tether length, the length of the flavin-dsDNA ligand was extended to 100 bp corresponding to almost 35 nm, i.e. five times the diameter of dodecin. For this purpose, the surface grafted ssDNA was subsequently hybridized with two oligomers of 80 bases and thereafter with flavin modified ssDNA as outlined in Scheme 1.The resulting flavin-dsDNA (100 bp) tether contains two nicks in the DNA backbone, which may introduce additional flexibility. As before, a solution of apododecin in buffer was incubated on the flavin-terminated dsDNA layer for a few minutes before the surface was rinsed with buffer solution. In Fig. 2 and S2† the corresponding QCM-D binding and unbinding curves are shown. On the surface with flavin-dsDNA (100 bp) tethers the adsorption of the apododecin considerably increases the dissipation-factor. Mechanical compliance of the extended DNA linkers results in the formation of a viscous layer, also indicated by spreading of the frequency overtones. The decrease in frequency upon apododecin adsorption is 2–3 times lower than for the 20 bp dsDNA tethers, which could be explained by a somewhat lower amount of flavin ligands at the surface, since the efficiencies of the individual hybridization steps leading to the formation of the flavin-dsDNA (100 bp) tethers are expected to be less than 100%. Additionally the formation of a less regular and less dense dodecin layer is expected, since with increasing tether length the reaching area of individual flavin-DNA tethers increases and for a single apododecin molecule more flavin ligands attached to the surface at different sites are available. To minimize the tether length, a double-stranded locked nucleic acid (dsLNA) tether formed by hybridization of flavin and 1,2-dithiane modified LNA was employed (see Fig. 3).
 | ||
| Fig. 2 Frequency (blue) and dissipation factor (red) shifts (overtones 5-13) observed in situ by QCM for adsorption/desorption of apododecin DtE on monolayers of flavin-terminated dsDNA with 100 bp as shown in the cartoon below. | ||
 | ||
| Fig. 3 Frequency (blue) and dissipation factor (red) shifts (overtones 5-13) observed in situ by QCM for adsorption/desorption of apododecin DtE on a monolayer of flavin-terminated dsLNA with 7 bp as shown in the cartoon below. | ||
In contrast to the typical B-type DNA/DNA helix, A-type conformation in DNA/LNA and LNA/LNA with a shorter base stacking distance and rigid duplex structure is expected to increase the thermal stability.26,27 Using LNA 7 bp were sufficient for strong hybridization at the surface leading to a long-term stable monolayer (the length of dsLNA with 7 bp is about 2 nm).28 Again a solution of apododecin in buffer was incubated for a few minutes before the surface was rinsed with buffer solution. Similar to the flavin-dsDNA tethers with 20 bp (see Fig. 1A) there is not much splitting of the frequency overtones in Fig. 3 and S3.† The fact that this time the overtones do not completely overlap can be explained by the formation of a less dense layer allowing some lateral motion of the adsorbed apododecin molecules. This assumption is supported by the decrease in frequency, which is 2–3 times less intense than for the flavin-terminated dsDNA layer with 20 bp. As the reaching area of the short flavin-LNA tethers is strongly decreased, less apododecin molecules can be captured.
As a consequence of the decreased reaching area of the tethers the probability for multi-ligand binding is less pronounced. This can also be deduced from the unbinding curve showing that by rinsing with buffer solution for a few minutes almost 80% of the apododecin molecules are being released. Again the spreading of the dissipation overtones indicates viscoelastic dispersion, which may originate from relaxation processes at rates comparable to the oscillation frequency of the resonator.
For the different tethers used in this study also the average distance between individual flavin-terminated oligonucleotide tethers is of interest. Due to the contribution of surface entrapped solvent molecules a determination of the increase in surface bound mass from the QCM-D data would not allow an accurate calculation of the surface coverage.23 Nevertheless the experimental data show that for the dsDNA and the ssDNA tethers used in Fig. 1 (with a length of about 7 nm) the distance between individual tethers is sufficiently short to allow the vast majority of the apododecin molecules to be captured by multi-ligand binding. As for the stepwise formation of the long flavin-dsDNA (100 bp) tethers (including three hybridization steps) the same type of capture probe layer as in Fig. 1A was used, but the hybridization efficiency for each step is expected to be less than 100%, the flavin surface coverage is expected to be somewhat lower than in Fig. 1A, but still sufficiently high to capture most apododecin molecules by multi-ligand binding. As for the flavin-dsLNA tethers with an overall length of 2–3 nm still about 20% of the adsorbed apododecin molecules are captured by multi-ligand binding, also here a rather dense layer was formed.
Repeatability of dodecin reconstitution on a flavin-terminated dsLNA monolayer
In order to prove whether it is still possible to release the apododecin molecules that remain captured at the flavin-terminated dsLNA monolayer also after rinsing due to multi-ligand binding by flavin reduction, a chemical reduction step consisting of rinsing the surface with an oxygen-free buffered solution of sodium dithionite (50 mM) was performed.1In Fig. 4 and S4† three times the adsorption of apododecin followed by rinsing with buffer solution and subsequent chemical flavin reduction resulting in the release of the remaining apododecin molecules is shown. In contrast to layers formed from flavin-terminated dsDNA of 20 bp the multiple binding and quantitative release of apododecin could only be achieved on the thin flavin-terminated dsLNA monolayer, if a fresh QCM crystal was applied (usually after a QCM experimental run the crystal can be cleaned and reused a few times). Apparently the quality of a monolayer formed by chemisorption of short LNA tethers is extremely sensitive against surface roughness, whereas minor inhomogeneities in the gold surface can be compensated by longer dsDNA tethers.
 | ||
| Fig. 4 Frequency (blue) and dissipation factor (red) shifts (overtones 5-13) observed in situ by QCM-D measurements. Three times the adsorption of apododecin DtE on monolayers of flavin-terminated dsLNA with 7 base pairs, followed by rinsing with buffer solution, and subsequently by rinsing with an oxygen-free buffered sodium dithionite solution for flavin reduction in order to release remaining DtE molecules captured by multi-ligand binding was measured. For the second and third chemical reduction step the period for rinsing with sodium dithionite had to be subsequently expanded. Prior to the first incubation of DtE a non-binding apododecin variant, W36A, was incubated as negative control at a concentration of 5 μM and subsequently rinsed with buffer to ensure that there was no unspecific binding.2,5 | ||
Determination of the binding parameters for flavin-ssDNA and dsDNA
To prove whether there is significant difference in the binding of flavin-ssDNA or dsDNA ligands SPFS adsorption/desorption studies of a fluorophore-labeled flavin-dsDNA ligand and a flavin-ssDNA ligand were carried out on a stable dodecin DtE monolayer as shown in Fig. 5. As negative control also the fluorescence signal caused by a non-binding (flavin-free) fluorophore-labeled ssDNA ligand was measured.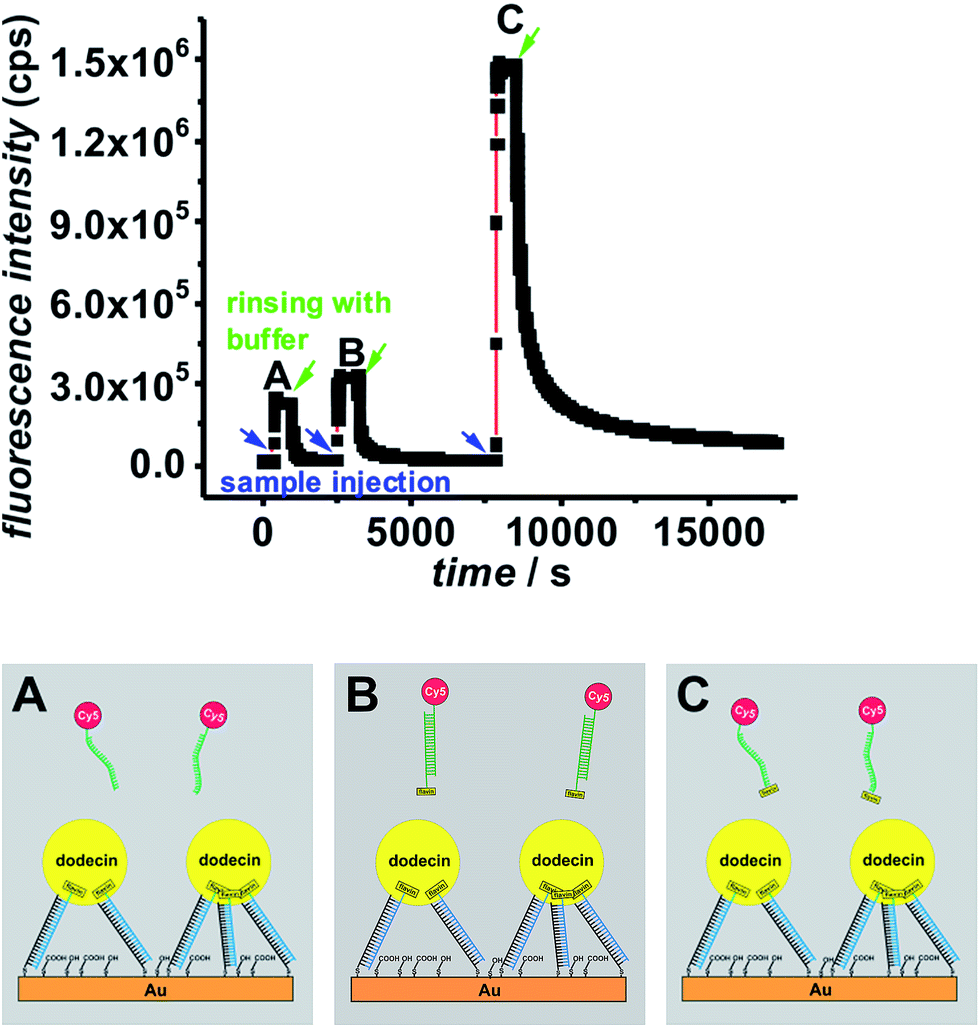 | ||
| Fig. 5 SPFS kinetic scan curve collected during ligand addition and subsequent rinsing with buffer (start of rinsing is indicated by arrows) for a Cy5-labeled non-binding (flavin-free) ssDNA ligand (A), a Cy5-labeled flavin-dsDNA ligand (B), and a Cy5-labeled flavin-ssDNA ligand (C) measured on the same dodecin-DNA-layer. | ||
After addition of the non-binding ligand (A) the fluorescence signal decreases as soon as the ligand containing solution is removed from the cell. During incubation of the dsDNA ligand (B) the increase in the fluorescence signal is only little larger than for the negative control, and the vast majority of this ligand could be removed after less than one hour of rinsing with buffer. In contrast for the ssDNA ligand (C) the increase in fluorescence is much larger indicating that a lot more ligand molecules are bound. The difference in fluorescence increase is so large that binding seems to follow faster kinetics and cannot only be explained by a larger amount of bound ligand molecules. In addition the unbinding kinetics of the ssDNA ligand are slower, since after rinsing for one hour still a significant amount of ligand is captured. For a detailed analysis of the unbinding kinetics the experimental curves were fitted exponentially as presented in the ESI, Fig. S6.† If the signal increase by fluorophores, which are present in the cell without being bound to the surface, is neglected, the unbinding kinetics of the dsDNA ligand can be fitted mono-exponentially, whereas two exponential functions are necessary in order to fit the unbinding kinetics of the ssDNA ligand. The latter finding can be explained by the fact that in principle each dodecin binding pocket can capture up to two ligands, and the second binding/unbinding event may follow different kinetics than the first. If this holds true for the ssDNA ligand, it can further be assumed that in the case of the dsDNA ligand only a single ligand is bound to each binding pocket. Probably the rather large and stiff dsDNA substructure acts as a stopper and prevents a second ligand from entering the binding pocket for steric reasons. Individual values of kon and koff obtained by fitting the SPFS unbinding kinetics exponentially are presented the ESI.† Since the accuracy of the analysis of the SPFS data is limited due to additional contribution of non-bound molecules to the fluorescence signal (as shown by the negative control), the corresponding experiments were carried out in the QCM-D cell as well, shown in Fig. 6 and S5.† Also these measurements show that a lot more molecules of the flavin-ssDNA can be captured than of the flavin-dsDNA (assuming there is not much difference in the number of trapped solvent molecules, which also contribute to the overall shift in frequency).23 A detailed analysis of the kinetic QCM binding/unbinding curves revealed that binding and unbinding of the flavin-dsDNA ligand can be fitted by a single exponential curve as presented in the ESI, Fig. S7.† Apparently only one flavin-dsDNA ligand can be captured in each binding pocket. Average values of kon = 1.3 × 103 M−1 s−1, koff = 3.5 × 10−3 s−1, and Kd = 2.7 × 10−6 M were determined from three experimental runs.
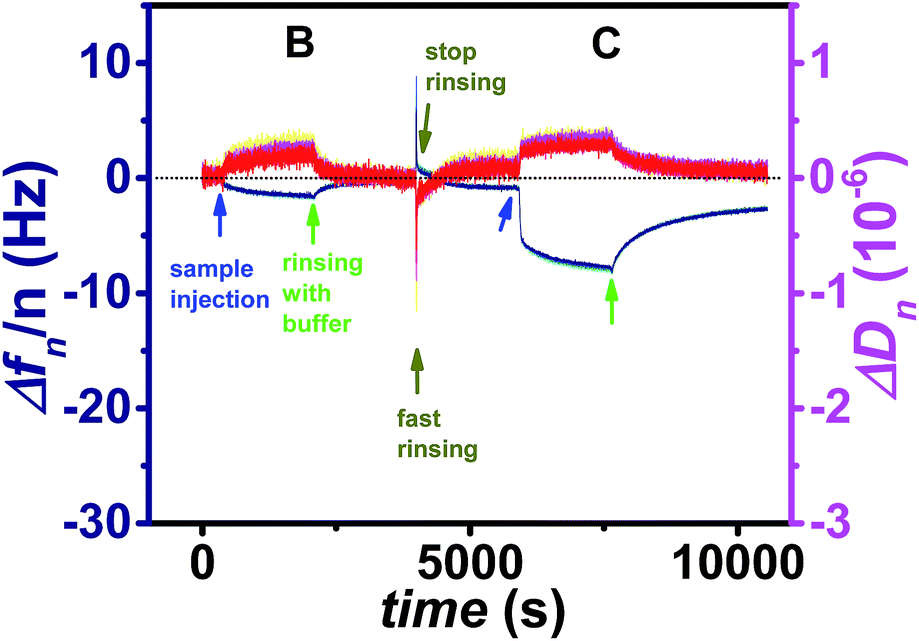 | ||
| Fig. 6 Frequency (blue) and dissipation factor (red) shifts (overtones 5-13) corresponding to the ligand addition and subsequent rinsing with buffer (start of rinsing is indicated by arrows) for a Cy5-labeled flavin-dsDNA ligand (B), and a Cy5-labeled flavin-ssDNA ligand (C). | ||
In accordance with the analysis of the SPFS unbinding curve, two exponential functions were required to fit the binding/unbinding curve of the flavin-ssDNA ligand in Fig. S7.† For the first binding/unbinding event, i.e. binding of the ligand to an empty and release from a singly occupied pocket average values of kon = 1.6 × 104 M−1 s−1, koff = 2.2 × 10−3 s−1, and Kd = 1.4 × 10−7 M were obtained. Apparently for binding to an empty binding pocket the flavin-ssDNA ligand binds with a kon value that is about one order of magnitude larger than for the flavin-dsDNA ligand, whereas both ligands follow nearly the same unbinding kinetics. While after binding a single flavin-dsDNA ligand the entrance to the pocket seems to be blocked, after binding a first flavin-ssDNA ligand a second flavin-ssDNA ligand may enter the same pocket following slower kinetics. For the second binding event a mean value of kon = 7.0 × 102 M−1 s−1 has been determined. The binding of the second ligand is expected to be stabilized by aromatic tetrade formation inside the binding pocket i.e. by π-stacking interaction between the two isoalloxazine moieties of the flavins and the two tryptophans W36 belonging to the apoprotein.2,5 This results in a relatively low value of koff = 1.9 × 10−4 s−1. Since for the second binding event not only kon but also koff is smaller than for the first, the value of Kd for the second binding/unbinding event (Kd = 2.7 × 10−7 M) is similar to that of the first (Kd = 1.4 × 10−7 M).
Conclusions
We have compared the nanomechanical properties of flavin-terminated oligonucleotide tethers of different length and composition for the formation of dodecin monolayers on gold. Our results show that most protein molecules can be captured leading to rather stiff layers with relatively high protein density, if the length of the tethers is in the same range as the diameter of dodecin. When significantly longer tethers (about five times the diameter of dodecin) are used, the DNA sublayer becomes heterogeneous and less protein molecules are captured leading to less dense and viscose layers. When on the other hand rather short flavin-dsLNA tethers (with a length of about one third of the diameter of dodecin) are used, the reaching area of individual tethers is too low to capture single apododecin molecules cooperatively, and the formation of stable and relatively dense protein layers is not possible. Nevertheless some of the absorbed protein molecules (about 20%) could be captured via multi-ligand binding. These molecules could be removed again by chemical flavin reduction, as shown previously for flavin-dsDNA tethers with 20 bp.1By comparing ssDNA and dsDNA it turned out that each dodecin binding pocket binds only a single flavin-dsDNA ligand whereas two flavin-ssDNA ligands can be incorporated. As the dsDNA is rather bulky and stiff, the kinetics for binding the dsDNA ligand to an empty pocket are slower than for the flavin-ssDNA ligand, but the unbinding kinetics are about the same. For the flavin-ssDNA ligands a second binding event is possible comprising smaller values for kon and koff than the first. As a consequence the binding strength reflected by the Kd values are about the same for the first and second binding/unbinding event of the flavin-ssDNA ligand, whereas the flavin-dsDNA ligand binds about one order of magnitude weaker.
However, also for the first binding event of the flavin-ssDNA ligand the kon value of kon = 1.6 × 104 M−1 s−1 is much lower than expected for diffusion-limited complex formation. For a diffusion controlled process, i.e. if each collision between apododecin and flavin ligand results in complex formation a value for kon in the order of 108 M−1 s−1 or even 109 M−1 s−1 would be expected. As a possible explanation the rather low kon values might be originated from the dodecin binding mode fitting the classic lock-and-key model of protein ligand interaction quite well. This assumption is made based on crystallographic data from apododecin (with empty pockets) and dodecin complexes with different flavin ligands, which show little to no difference between Cα-atom positions.2–5 Thus the (apo)dodecin binding pockets comprise a fixed steric configuration and there is nearly no structural rearrangement of the complex upon ligand binding. Ligands can enter only if they approach the binding pocket in a precise orientation. For flavin-oligonucleotide ligands with larger flexibility there will be a (somewhat) higher probability for the isoalloxazine moiety to reach the final binding site, i.e. binding is favored, if a rather long and flexible linker in between isoalloxazine and oligonucleotide is present, and/or if the oligonucleotide as such is flexible, which is the case for ssDNA but not for dsDNA. This is in contrast to ligand binding by an induced fit mechanism that has been described e.g. for the prominent multi-ligand binding (apo)streptavidin–biotin system, which comprises nearly a diffusion-limited rate for complex formation.29,30 For streptavidin–biotin a value of kon ≈ 3.8 × 108 M−1 s−1 can be calculated from Kd ≈ 10 fM and koff = 3.8 × 10−6 s−1.29–35 Binding a biotin ligand is going along with mayor structural rearrangement, and one surface loop folds over the binding site when biotin is bound.36–39 Accordingly the rather low kon value of dodecin can be explained by its ligand binding mechanism. Taking the structural peculiarity of dodecin into account, a strategy for the formation of stable and dense protein layers with high mechanical stiffness could be the hybridization of a 20 bases capture probe strand (as used previously, e.g. as shown in Fig. 1A) with a flavin-modified complementary strand comprising 20 bases complementary to the capture probe, and an overhang of five to ten bases (e.g. thymidines) at the flavin-terminated 5′-end providing the flavin at a flexible subunit of ssDNA. In principle also flavin and thiol (or dithiane) modified ssDNA can be used (as shown in Fig. 1B), but the synthesis of ssDNA modified at both ends is synthetically more demanding. Furthermore it has been reported that the isoalloxazine moiety of flavins may absorb irreversibly at bare gold surfaces.40
This can be prevented if the gold surface is first modified with (flavin-free) ssDNA, followed by adsorption of short thiol molecules, before the flavin moiety is introduced in a hybridization step. This surface modification strategy has also the advantage that the flavin density at the surface (relative to the number of dsDNA-strands) can be varied by hybridization with mixtures of flavin-modified and flavin-free complementary ssDNA with different ratio as shown before.1
Acknowledgements
We thank Dr Roman Sheparovych for experimental support. This work has received funding from the European Research Council under the European Community's Seventh Framework Programme (FP7/2007-2013)/ERC Grant agreement no. 240544, from the country North Rhine-Westphalia, and the University of Siegen.References
- C. Gutiérrez Sánchez, Q. Su, H. Schönherr, M. Grininger and G. Nöll, ACS Nano, 2015, 9, 3491–3500 CrossRef PubMed.
- M. Grininger, G. Nöll, S. Trawöger, E.-K. Sinner and D. Oesterhelt, Biointerphases, 2008, 3, 51–58 CrossRef CAS PubMed.
- M. Grininger, F. Seiler, K. Zeth and D. Oesterhelt, J. Mol. Biol., 2006, 364, 561–566 CrossRef CAS PubMed.
- M. Grininger, K. Zeth and D. Oesterhelt, J. Mol. Biol., 2006, 357, 842–857 CrossRef CAS PubMed.
- Y. Yu, B. Heidel, L. Parapugna Tamara, S. Wenderhold-Reeb, B. Song, H. Schönherr, M. Grininger and G. Nöll, Angew. Chem., Int. Ed., 2013, 52, 4950–4953 CrossRef CAS PubMed.
- M. Grininger, H. Staudt, P. Johansson, J. Wachtveitl and D. Oesterhelt, J. Biol. Chem., 2009, 284, 13068–13076 CrossRef CAS PubMed.
- G. Nöll, S. Trawöger, M. von Sanden-Flohe, B. Dick and M. Grininger, ChemBioChem, 2009, 10, 834–837 CrossRef PubMed.
- H. Staudt, D. Oesterhelt, M. Grininger and J. Wachtveitl, J. Biol. Chem., 2012, 287, 17637–17644 CrossRef CAS PubMed.
- T. Nöll and G. Nöll, Chem. Soc. Rev., 2011, 40, 3564–3576 RSC.
- C. Jeppesen, J. Y. Wong, T. L. Kuhl, J. N. Israelachvili, N. Mullah, S. Zalipsky and C. M. Marques, Science, 2001, 293, 465–468 CrossRef CAS PubMed.
- J. Y. Wong, T. L. Kuhl, J. N. Israelachvili, N. Mullah and S. Zalipsky, Science, 1997, 275, 820–822 CrossRef CAS PubMed.
- A. G. Moreira and C. M. Marques, J. Chem. Phys., 2004, 120, 6229–6237 CrossRef CAS PubMed.
- N. W. Moore and T. L. Kuhl, Biophys. J., 2006, 91, 1675–1687 CrossRef CAS PubMed.
- L. Feng, L.-L. Pontani, R. Dreyfus, P. Chaikin and J. Brujic, Soft Matter, 2013, 9, 9816–9823 RSC.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 2985–2993 CrossRef CAS.
- E. L. S. Wong, E. Chow and J. J. Gooding, Langmuir, 2005, 21, 6957–6965 CrossRef CAS PubMed.
- J. F. Marko and E. D. Siggia, Macromolecules, 1995, 28, 8759–8770 CrossRef CAS.
- R. Vafabakhsh and T. Ha, Science, 2012, 337, 1097–1101 CrossRef CAS PubMed.
- T. Nöll, H. Schönherr, D. Wesner, M. Schopferer, T. Paululat and G. Nöll, Angew. Chem., Int. Ed., 2014, 53, 8328–8332 CrossRef PubMed.
- B. Tinland, A. Pluen, J. Sturm and G. Weill, Macromolecules, 1997, 30, 5763–5765 CrossRef CAS.
- D. Johannsmann, I. Reviakine and R. P. Richter, Anal. Chem., 2009, 81, 8167–8176 CrossRef CAS PubMed.
- F. Höök and B. Kasemo, in Piezoelectric sensors, ed. C. Steinem and A. Janshoff, Springer, 2007, pp. 425–447 Search PubMed.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- D. Johannsmann, I. Reviakine, E. Rojas and M. Gallego, Anal. Chem., 2008, 80, 8891–8899 CrossRef CAS PubMed.
- D. Johannsmann, in The Quartz Crystal Microbalance in Soft Matter Research, Springer, 2015, pp. 33–47 Search PubMed.
- A. K. Thazhathveetil, J. Vura-Weis, A. Trifonov, M. R. Wasielewski and F. D. Lewis, J. Am. Chem. Soc., 2012, 134, 16434–16440 CrossRef CAS PubMed.
- D. A. Braasch and D. R. Corey, Chem. Biol., 2001, 8, 1–7 CrossRef CAS PubMed.
- A. Eichert, K. Behling, C. Betzel, V. A. Erdmann, J. P. Fürste and C. Förster, Nucleic Acids Res., 2010, 38, 6729–6736 CrossRef CAS PubMed.
- V. H. Pérez-Luna, M. J. O'Brien, K. A. Opperman, P. D. Hampton, G. P. López, L. A. Klumb and P. S. Stayton, J. Am. Chem. Soc., 1999, 121, 6469–6478 CrossRef.
- L. S. Jung, K. E. Nelson, P. S. Stayton and C. T. Campbell, Langmuir, 2000, 16, 9421–9432 CrossRef CAS.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 1200–1216 CrossRef CAS PubMed.
- O. H. Laitinen, V. P. Hytonen, H. R. Nordlund and M. S. Kulomaa, Cell. Mol. Life Sci., 2006, 63, 2992–3017 CrossRef CAS PubMed.
- C. Grunwald, Z. Phys. Chem., 2008, 222, 789–821 CrossRef CAS.
- W. Müller, H. Ringsdorf, E. Rump, G. Wildburg, X. Zhang, L. Angermaier, W. Knoll, M. Liley and J. Spinke, Science, 1993, 262, 1706–1708 Search PubMed.
- X. Su, Y.-J. Wu, R. Robelek and W. Knoll, Langmuir, 2005, 21, 348–353 CrossRef CAS PubMed.
- S. Freitag, I. Le Trong, L. Klumb, P. S. Stayton and R. E. Stenkamp, Protein Sci., 1997, 6, 1157–1166 CrossRef CAS PubMed.
- P. C. Weber, D. Ohlendorf, J. Wendoloski and F. Salemme, Science, 1989, 243, 85–88 CAS.
- O. Livnah, E. A. Bayer, M. Wilchek and J. L. Sussman, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5076–5080 CrossRef CAS.
- I. P. Korndörfer and A. Skerra, Protein Sci., 2002, 11, 883–893 CrossRef PubMed.
- G. Nöll, E. Kozma, R. Grandori, J. Carey, T. Schödl, G. Hauska and J. Daub, Langmuir, 2006, 22, 2378–2383 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The different presentation of QCM-D readout is shown and the analysis of kinetics is presented. See DOI: 10.1039/c6ra10090b |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |