
Evaluation of the catalytic effect of non-noble bismuth on the lead half-cell reaction for lead-based redox flow batteries
Zhaolin Naab,
Fei Lianga,
Dongming Yinab and
Limin Wang*ac
aState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, CAS, Changchun, 130022, China. E-mail: lmwang@ciac.ac.cn; Fax: +86-0431-85262836; Tel: +86-0431-85262447
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cChangzhou Institute of Energy Storage Materials and Devices, Changzhou, 213000, P. R. China
First published on 6th June 2016
Abstract
Bi3+ ions are introduced into the lead electrolyte of lead-based redox flow batteries (RFBs), and their influence on the electrochemical performance of the redox cell is thoroughly investigated. The metallic Bi particles, which are simultaneously electrodeposited onto the electrode surface during the charge process of the RFBs, significantly improve the electrochemical performance of lead-based RFBs by enhancing the activity and reversibility of the sluggish Pb(II)/Pb(0) redox reaction and suppressing hydrogen evolution. The cell using a negative electrolyte with Bi3+ ions exhibits considerable enhancement both in the columbic efficiency (CE) and voltage efficiency (VE), and therefore in the energy efficiency (EE). Moreover, a mechanism accounting for the role that Bi particles play in the redox reactions in this lead half-cell is proposed. Bi particles favor the formation of a BiHx compound, an intermediate that reduces Pb(II) to Pb(0), and thereby curbs the competitive side reaction of hydrogen evolution responsible for the major loss for the CE. Additionally, the morphology of lead electrodeposition is also presented and the deposits become more uniform and smooth without any dendrites upon the addition of Bi3+. The results suggest that the utilization of non-noble Bi as a high-performance additive promises to be applicable to lead-based RFBs.
Introduction
Large scale energy storage is a potentially important and cost-effective approach to enhance the overall efficiency and power quality of the grid by effectively balancing the mismatch between supply and demand. This may become more important when a great amount of energy produced from intermittent and fluctuating renewable power sources is integrated with the electrical grid in the context of global climate change and limited fossil fuel reserves. Redox flow batteries (RFBs) are considered one of the most promising energy storage technologies for large-scale stationary energy storage, especially for high energy applications.1,2 They offer several key advantages over enclosed batteries (e.g., Li-ion, lead-acid) including modular design, simplified manufacturing, long service life and moderate maintenance cost.3–5 RFB systems typically possess a unique configuration that consists of three key components: cells stacks, flow system, and electrolyte storage tanks.6 Unlike the enclosed batteries where energy is stored within solid electrodes in a single vessel, RFBs typically employ soluble active species contained in the liquid electrolytes which are stored in the tanks external to the battery.7 Upon operating, the catholyte and anolyte containing active species are driven by the pump and circulate through their respective half-cells. The chemical energy in electrolytes is reversibly converted to electrical energy as the electrolytes are flowing through the electrodes.8 Thanks for such a unique configuration, RFBs possess the most attractive feature that other battery systems do not have: decoupled energy and power, which enables an independent control of capacity and power to meet different energy storage requirements in a wide range of a few kW h to several MW h.9Since the 1970s, a large number of RFB systems have been developed, such as iron-chromium,10,11 zinc-bromine,12 all-vanadium,13–17 and soluble lead-acid.18,19 Among the different types of RFBs, the all-vanadium RFB system is the most developed system mainly owing to its reversibility and reduced cross-contamination by using the same element. Despite these advantages, relatively low cell voltage (c.a. 1.26 V) and energy density (c.a. 20–30 Wh L−1) remain the primary research challenges of this RFB system, which have been highlighted in several recent reports.20 Recent developments in RFB research have shown increasing interesting in redox couple combinations which deliver high cell voltage and thereby high energy density systems.21 Among them the cerium couple Ce(III)/Ce(IV) offering a high positive potential of 1.72 V vs. NHE22 has been studied in several solvent compositions.20,23 And one of the most studied is the cerium-zinc RFB firstly developed in 2004 by Plurion Inc.24–30 In a recent paper,31 we reported preliminary studies of a novel cerium-lead RFB system where the electrode reactions are:
As for the positive half-cell

While in the negative half-cell
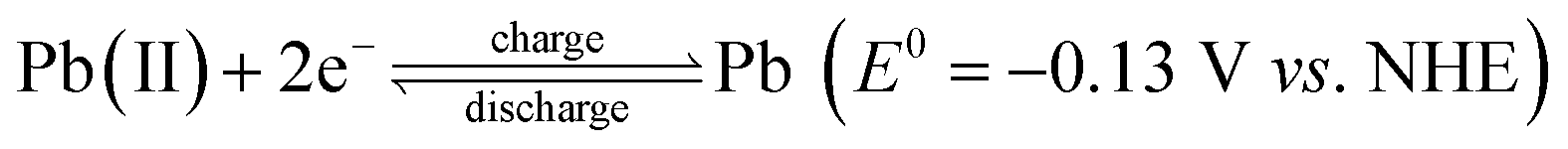
The overall cell reaction

As for the charging process, Ce(III) are oxidized to Ce(IV) ions at the positive half-cell, while electrodeposition of lead takes place at the negative electrode. Upon discharge, the inverse reactions take place, where the anodically generated Ce(IV) ions in the positive electrolyte are reduced back to Ce(III) ions accompanied by oxidation and redissolution of the electrodeposited lead at the negative electrode. Methanesulfonic acid (MSA) serves as supporting electrolyte in the both positive and negative half-cell, as these electrolytes enable high concentrations of highly electropositive lead and cerium ions.28 In addition, MSA is a strong sulfonic acid with lower vapor pressure and less corrosive than other common supporting electrolytes (e.g., hydrochloric and sulfuric acids).32
The cerium-lead RFB is attractive for several reasons. Its thermodynamic cell potential (1.85 V) is among the highest of aqueous RFBs. Moreover, the required reagents with moderate cost do not pose the risk of toxic halide emissions while possessing significantly lower toxicity than vanadium. Additionally, the flow cell can be operated through more than 800 charge/discharge cycles and in the broad temperature range between 253 and 313 K. However, the practical utilization of the cerium-lead RFB system is hampered by its low energy efficiency (EE). Therefore, prior to its further possible commercialization, the EE of this system should be enhanced. Hydrogen evolution in the last stage of charge in the lead half-cell is one of the reasons leading to the decrease of the coulombic efficiency (CE) and so the EE.18 Moreover, dendritic growth of the lead electrode has been a major problem in the lead-based RFBs that often does not allow a thick electrodeposit, which further limits the real EE.19 Hence, practical applications of cerium-lead RFBs are limited by above problems of the lead half-cell.
In the electroplating and battery industries, electrolytic additives are usually used to obtain more uniform and compact deposited coatings, and to minimize undesired side reactions. For instance, some inorganic or organic additives (e.g. indium oxide and tetrabutylammonium hydroxide) have been added into zinc electrolyte in order to facilitate zinc electrodeposition.33 In the case of lead deposition, additives that could enhance the quality of the lead deposit morphology, increase the kinetics of the lead electrode and inhibit hydrogen evolution, are urgently to be developed and evaluated. Herein, we report a non-noble and highly-conductive metallic Bi as a novel catalyst to enhance the electrochemical activity and reversibility of the Pb(II)/Pb(0) redox reaction, enabling the RFBs for high energy efficiency operation. Briefly, an appropriate amount of Bi3+ ions was added to the lead electrolytes directly, and the metallic Bi particles are then simultaneously reduced and electrodeposited on the electrode surfaces during the operation of the RFBs, without any complex pretreatment procedures. And the catalytic effects of Bi on the lead electrodeposition and electrochemical performance of the cell are investigated. What is more, the detailed discussion of system efficiency and lead morphology is included in this work.
Experimental
All the chemicals were used as received without further purification and the solutions were prepared with deionized water. The electrolytes were purged with a rapid stream of N2 bubbles for 30 min to avoid interference from the oxygen. Pb(II) methanesulfonate solutions were produced by dissolving Pb(II) oxide (Aladdin, 99.9 wt%) into reagent grade aqueous MSA (Alfa Aesar, 70 vol%) under magnetic stirring. The electrolyte with the Bi3+ ions was prepared by adding Bi2O3 (Aladdin, 99.9 wt%) into above solutions.Electrochemical measurements were performed in a three-electrode electrochemical cell, with a graphite plate (area: 1.0 cm2) as the working electrode, a saturated Ag/AgCl electrode as the reference electrode, and a large area graphite sheet as the counter electrode. The graphite plate electrodes were polished with 800 and 1500 grit SiC paper, and then washed with deionized water and ethanol before experiments. Cyclic voltammograms were recorded in electrolytes including 0.05 M Pb(II) methanesulfonate + 1.0 M MSA with and without 4 mM Bi3+ ions. Linear sweep voltammetry (LSV) experiments were carried out in a 1.0 M MSA with or without 4 mM Bi3+ ions solution. The initial potential was 0.4 V and the final potential is −1.35 V at 10 mV s−1 potential sweep rate. The electrochemical measurements were conducted on a VMP3 electrochemical workstation (Bio-Logic).
The charge/discharge tests were carried out in an in-house designed single static cell. Details of the cell setup were depicted in a previous article.31 Briefly, a commercially available low-cost cation-exchange membrane (GEFC-104, Golden Energy) separated the two compartments. Graphite plate with an exposed electrode area of 4 cm × 2 cm (8 cm2) was used for both the negative and the positive electrodes. Prior to each experiment, the graphite electrodes were mechanically polished and then cleaned in an ultrasonic bath for 5 min in a deionized water/ethanol mixture. The negative electrode compartment contained 15 mL of 1.5 M Pb(II) methanesulfonate in 1.0 M MSA with and without 4 mM Bi3+ while the positive electrode compartment contained 15 mL of 1.0 M MSA. The cells were charged at a constant current density of 20 mA cm−2 (160 mA) for 2 h and discharged until the voltage dropped down to 0.5 V at the same current density (20 mA cm−2) under ambient temperature. The charge/discharge measurements were made by a BTS battery test system (NEWWARE, 5 V/1A).
The scanning electron microscope (SEM) images and energy-dispersive X-ray spectroscopy (EDS) element mapping of the lead electrodeposits were obtained on a Hitachi S-4800 field emission scanning electron microscope using an acceleration voltage of 10 kV. Prior to taking the microscopic images, the samples were rinsed with deionized water, and then dried for 5 h at 60 °C.
Results and discussion
The CV curves for Pb electrodeposition measured between −0.7 V and 0.4 V at a scan rate of 10 mV s−1 in the pristine electrolyte (0.05 M Pb(II) methanesulfonate + 1.0 M MSA) and the electrolyte with 4 mM Bi3+ ion are shown in Fig. 1. The cathodic and anodic sweeps can be seen as the charge and discharge cycles in a lead half-cell, respectively. It can be clearly seen that both curves exhibit two main peaks, which is assigned to the reduction of Pb(II) to Pb(0), and the stripping of the deposited Pb. For the CV curves of the electrolyte with Bi3+ ion, a smaller reduction peak at c.a. −0.07 V prior to the main cathodic peak can be observed, which corresponds to Bi(III) reduction. While, there are also two small oxidation peaks at around 0.1 and 0.13 V, following the first anodic peak and associated with the oxidation of Bi.34 As observed, the peak potential difference decreases and the peak current increases after the addition of Bi3+, suggesting that this additive could improve the electrochemical activity and reversibility of the Pb(II)/Pb(0) redox reaction in the negative electrolyte of RFB. And the figure also shows that the onset reduction peak shifts positively in the presence of Bi3+, implying that the electrodeposition reaction of lead with the introduction of the additive requires a smaller overpotential. Another significant difference between the two CV profiles is that a significant nucleation loop is observed for the sample without additive but not with the addition of Bi3+ ion. The appearing of a nucleation loop is attributed to the fact that the nucleation and growth can still occur during the reverse scan until the anodic stripping potential is reached.35 This behavior (nucleation loop) further indicates that the deposition of Pb without additive needs more overpotential than that with Bi additive. The above results demonstrate that the introduction of Bi3+ ions can facilitate the redox reaction between Pb(II) and Pb(0), which owns to the electrocatalytic effect of Bi. Since the standard potential of Bi(III)/Bi(0) (0.046 V vs. Ag/AgCl)36 is more positive than that of Pb(II)/Pb(0), metallic Bi will be electrodeposited on the electrode surface prior to the reduction of Pb(II) to Pb(0). Therefore, it is metallic Bi rather than Bi3+ ion that has catalytic effect toward lead electrodeposition and Bi presents in the form of metallic Bi in the lead half-cell. | ||
| Fig. 1 CVs performed in the electrolytes of 0.05 M Pb(II) methanesulfonate + 1.0 M MSA with or without 4 mM Bi3+ from 0.2 V to −0.7 V at a scan rate of 10 mV s−1. | ||
To further investigate the effects of the Bi3+ on the electrochemical performance, typical series of CV curves of the lead electrolyte (0.05 M Pb(II) methanesulfonate + 1.0 M MSA) with and without Bi3+ ions at a range of scan rates are shown in Fig. 2. The peak currents and potential interval of this two measured electrolyte increase with the augment of potential sweep rate, as shown in Fig. 2, which indicates that the Pb(II)/Pb(0) redox reactions in these solutions are quasi-reversible. As for the quasi-reversible nature of the Pb(II)/Pb(0) redox reaction, the relationship between the anodic peak current and the scan rate can be predicted by the Randles–Sevcik equation.37
 | ||
| Fig. 2 CV curves of (a) the pristine electrolyte (0.05 M Pb(II) methanesulfonate + 1.0 M MSA) and (b) the pristine electrolyte with 4 mM Bi3+ at different scan rates. | ||
For the reversible reaction,
| ip = 2.69 × 105n3/2ACD01/2υ1/2 | (1) |
For the irreversible reaction,
| ip = 2.99 × 105n3/2α1/2ACD01/2υ1/2 | (2) |
In this case, as mentioned above, the Pb(II)/Pb(0) redox reactions with or without the Bi3+ are probably quasi-reversible. Hence the diffusion coefficient (D0) of the Pb(II) species in these electrolytes should fall between the D0 of Pb(II) species for a reversible Pb(II)/Pb(0) redox reaction and that for an irreversible one. Fig. 3 illustrates the plots of the cathodic peak current versus the square root of the sweep rate in different solutions. Through the slope of the linear fitting to the plot of ip vs. υ1/2, the D0 of Pb(II) species for a reversible or an irreversible redox reaction could be calculated. The D0 of Pb(II) ions with the addition of Bi3+ is found to be 1.69–2.74 × 10−5 cm2 s−1, while that of the pristine electrolyte is 5.08–8.22 × 10−6 cm2 s−1. Obviously, the diffusion coefficient for Pb(II) ions increases with the addition of Bi3+ ion. And this could be another reason for the enhanced electrochemical activity and reversibility of the Pb(II)/Pb(0) redox reaction after the introduction of Bi3+ ion.
 | ||
| Fig. 3 Plots of the cathodic peak current (ip) vs. the square root of scan rate (υ1/2) for the pristine electrolyte (0.05 M Pb(II) methanesulfonate + 1.0 M MSA) with and without 4 mM Bi3+. The straight lines are linear fittings to the plots (ip vs. υ1/2). | ||
Fig. 4 shows up to 100th repetitive CV curves measured in the electrolyte with 4 mM Bi3+ ion at a scan rate of 50 mV s−1. The curve shape and peak location almost have no change after 100 cycles. This implies that the lead electrolyte with Bi3+ ion as an additive possesses good cycling stability.
 | ||
| Fig. 4 CV curves of the electrolyte (0.05 M Pb(II) methanesulfonate + 1.0 M MSA) with 4 mM Bi3+ during the 1st and 100th cycles from 0.4 V to −0.7 V at 50 mV s−1. | ||
Aiming to further study the catalytic effect of Bi particles on the electrochemical performance of the lead half-cell, the charge–discharge experiments are carried out in a single static cell. The electrolyte composition was 1.5 M Pb methanesulfonate in 1.0 M MSA with and without 4 mM Bi3+ in the negative electrode compartment while the positive electrode compartment contains 1.0 M MSA. For comparison, Fig. 5a shows the typical charge–discharge profiles for the cell at the same charge/discharge current density of 20 mA cm−2 with the pristine negative electrolyte and the electrolyte containing 4 mM Bi3+. It can be seen that the cell using electrolyte containing Bi3+ exhibits lower charge voltage plateau and higher discharge voltage plateau compared with the pristine one. This result indicates that the introduction of Bi3+ to the negative electrolyte could enhance electrochemical activity and kinetic reversibility of the electrochemical reactions in the electrolyte and thereby reduce the overpotential. This is consistent with the results of the CV and diffusion coefficient measurements.
 | ||
| Fig. 5 (a) Charge/discharge curves, (b) CEs, (c) VEs and (d) EEs measured for the cell employing the pristine negative electrolyte (1.5 M Pb(II) methanesulfonate + 1.0 M MSA) with and without 4 mM Bi3+. Charge at 20 mA cm−2 for 2 h and discharge at 20 mA cm−2 until to 0.5 V. | ||
The coulombic efficiencies (CEs), voltage efficiencies (VEs) and energy efficiencies (EEs) of the cell using the negative electrolyte with and without Bi3+ as a function of cycling number are shown in Fig. 5b–d, respectively. With the addition of Bi3+, the energy efficiency improved significantly from 48% without additive to 61%. In this case, higher CE and EE are obtained simultaneously. As presented in the voltammetry study, improvement of the electrochemical activity and reversibility of the electrolyte with Bi3+ was observed. Therefore, a higher VE is expected in this study. The enhancement in CE is believed to be a result of the presence of Bi acting as efficient inhibiter to reduce the side reaction of hydrogen evolution. It has been reported that Bi could increase the hydrogen evolution overpotential in some circumstances.38 To verify this hypothesis in this case, linear sweep voltammograms (LSVs) in the presence and absence of additives are carried out in an electrolyte containing 1.0 M MSA without lead salts.
Fig. 6 shows LSV curves obtained for these two samples under study. Contrary to expectation, the redox overpotential of hydrogen evolution in the presence of Bi3+ ions is lower than that of pristine electrolyte. What is more, in the potential range where the negative lead half-cell works (inset of Fig. 6), the currents measured corresponding to the sample with Bi3+ is clearly higher than that of the pristine one. These results indicate that the enhanced CE of the cell using the electrolyte with Bi3+ probably does not stem from the increase in the hydrogen evolution overpotential. Herein, a more complex redox action mechanism involving the lead salts is proposed to explain the enhancement in CE of the cell using the electrolyte with the additive (Fig. 7). In the absence of the additive, the graphite electrode can be considered as a single surface (carbon) that can provide active sites for the two parallel redox reactions during the charge process. The main redox process is the reduction of Pb2+ to Pb [Fig. 7a, eqn (1)], accompanied by the competitive reaction of hydrogen evolution that can also takes place at such very negative potentials [Fig. 7a, eqn (2)]. This latter reaction is probably favored by carbon surfaces having little oxygen-containing functional group. As for the case with Bi3+ (Fig. 7b), metallic Bi particles are reduced and deposited on the surface of electrodes prior to the lead electrodeposition. So the electrodes can be considered as a system with two different surfaces (a carbon surface and a metallic Bi surface) which compete as active sites for the two parallel redox reactions during the charge process. The lead electrodeposition may take place mainly on the carbon surface and the major difference between the two samples is found to be in the reduction process of H+ ions. Several reports have demonstrated the formation of BiH3 (ref. 39) or nonstoichiometric BiHx compounds40 when metallic Bi electrodes are subjected to very negative potentials [Fig. 7b, eqn (3)]. These compounds are formed as previous intermediates to the evolution of hydrogen. And the BiHx has also been confirmed to have strong reduction ability and even can reduce a layer of Bi2O3 to Bi.39 In this system we propose that BiHx compound reacts with the Pb2+ ions from the electrolyte to form metallic Pb, rather than decompose to release hydrogen [Fig. 7b, eqn (4)]. Thus, the overall redox reaction taking place at the Bi particles should be the reduction of Pb2+ to Pb(0) [Fig. 7b, eqn (5)]. Consequently, the total charge consumed during the charge process on this electrode is used directly or indirectly for lead electrodeposition. And so the irreversible side reaction of hydrogen evolution is inhibited, which could contribute to the enhancement in CE.
 | ||
| Fig. 6 LSVs performed in the electrolytes of 1.0 M MSA with and without 4 mM Bi3+ at 10 mV s−1. | ||
 | ||
| Fig. 7 Scheme of the redox reactions occurring in the lead negative half-cell: (a) on a carbon surface and (b) in the presence of metallic Bi. | ||
The dendritic nature of deposited lead coatings, leading to short circuit and therefore reduced cycle life, also requires careful consideration in order to maximize the efficiency of the cell. Thus, the electrolytic additive of Bi3+ ions for dendrite suppression is evaluated. The morphology of the lead electrodeposits is studied by scanning electron microscopy (SEM). Fig. 8 shows the obtained lead coatings after the cell is charged at 20 mA cm−2 for 2 h. It has been noted that evolution of dendritic growth tends to concentrate typically at the edges of electrodes where the local overpotential is higher compared to that in the center regions.41 So these SEM images have focused on the edges of the electrode surface. It can be seen that the obtained deposits from pristine electrolytes are uniform and dendritic, and have an irregular form of different sizes (Fig. 8a). This dendritic growth suggests non-uniform current and potential distribution at the electrode surface resulting in a rough deposit that can be easily dislodged. By contrast, in the presence of Bi3+, the deposits obtained are more regular and smooth with no evidence of dendritic growth (Fig. 8b). The corresponding EDS mapping analysis of the coating is displayed in Fig. 8c–f. It can be seen that Pb and Bi elements are co-existent and show homogeneous dispersion within the deposits. Therefore, Bi3+ ions are attractive for lead-based RFBs, as they optimize the surface morphology of the lead deposits, which can not only minimize the short circuit but also avoid the loss of deposited material in the cell and so enhance the CE.
 | ||
| Fig. 8 SEM images of the lead electrodeposits obtained from the charge/discharge experiments of the cell in (a) the pristine negative electrolyte (1.5 M Pb(II) methanesulfonate + 1.0 M MSA) electrolytes and (b) the pristine electrolyte with 4 mM Bi3+. (c–f) Corresponding EDS mapping of the lead electrodeposit obtained from the negative electrolyte with 4 mM Bi3+. Charge for 2 h at 20 mA cm−2. | ||
Conclusions
In conclusion, Bi particles acting as a low-cost, conductive, and highly-effective electrocatalyst toward the lead electrodeposition reaction is proposed to enhance the electrochemical performance of lead half-cell in lead-based RFB systems. The metallic Bi particles are synchronously electrodeposited on the electrode surface as catalysts in the lead half-cell when the cells are operated with the electrolytes containing Bi3+ ions. This Bi particles deposited on the surfaces of electrodes could enhance the electrochemical activity and reversibility of the Pb(II)/Pb(0) redox reaction thus leading to greatly increased cell performance in VE. Furthermore, a mechanism that explains the role of Bi particles in the presence of lead salts has been proposed. Bi particles favor the formation of BiHx, an intermediate that reduces Pb(II) to Pb(0), and therefore inhibits the competitive irreversible hydrogen evolution. Consequently, the total charge consumed during the cathodic sweep on this electrode is used for the reduction of Pb(II) to Pb(0), resulting in an improved CE. Thus, the EE improved significantly from 48% in the absence of additives to 61% due to the improvement of both VE and CE in the presence of Bi. In addition, the morphology of lead deposits showed that the obtained electrodeposits became more regular and smooth with no evidence of dendritic growth in the presence of Bi. These results manifest that Bi3+ ions hold great promise as a high performance additive for lead-based RFB applications.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21373198, 21221061), and Natural Science Foundation of Jiangsu Province (BK20141174).Notes and references
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- K. X. Lin, Q. Chen, M. R. Gerhardt, L. C. Tong, S. B. Kim and L. Eisenach, Science, 2015, 349, 1529–1532 CrossRef CAS PubMed.
- G. P. Rajarathnam, M. E. Easton, M. Schneider, A. F. Masters, T. Maschmeyer and A. M. Vassallo, RSC Adv., 2016, 6, 27788–27797 RSC.
- M. Ulaganathan, V. Aravindan, Q. Y. Yan, S. Madhavi, M. Skyllas-Kazacos and T. M. Lim, Adv. Mater. Interfaces, 2016, 3, 1500309 CrossRef.
- S. S. Peng, X. M. Yan, D. S. Zhang, X. M. Wu, Y. L. Luo and G. H. He, RSC Adv., 2016, 6, 23479–23488 RSC.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970–986 CrossRef CAS.
- C. H. Wang, X. F. Li, X. L. Xi, P. C. Xu, Q. Z. Lai and H. M. Zhang, RSC Adv., 2016, 6, 40169–40174 RSC.
- X. S. Zhao, Y. Z. Fu, W. Li and A. Manthiram, RSC Adv., 2012, 2, 5554–5556 RSC.
- C. Ponce-de-León, A. Frías-Ferrer, J. González-García, D. A. Szánto and F. C. Walsh, J. Power Sources, 2006, 160, 716–732 CrossRef.
- L. H. Thaller, US Pat., 3996064, 1974.
- M. Lopez-Atalaya, G. Codina, J. R. Perez, J. L. Vazquez and A. Aldaz, J. Power Sources, 1992, 39, 147–154 CrossRef CAS.
- S. Suresh, T. Kesavan, Y. Munaiah, I. Arulraj, S. Dheenadayalan and P. Ragupathy, RSC Adv., 2014, 4, 37947–37953 RSC.
- Y. F. Wang, S. J. Wang, M. Xiao, D. M. Han, M. A. Hickner and Y. Z. Meng, RSC Adv., 2013, 3, 15467–15474 RSC.
- C. Ding, X. Ni, X. F. Li, X. L. Xi, X. W. Han, X. H. Bao and H. M. Zhang, Electrochim. Acta, 2015, 164, 307–314 CrossRef CAS.
- Y. Y. Zhao, M. R. Li, Z. Z. Yuan, X. F. Li, H. M. Zhang and I. F. J. Vankelecom, Adv. Funct. Mater., 2016, 26, 210–218 CrossRef CAS.
- W. X. Xu, H. M. Zhang, F. Xing, H. Z. Zhang, Y. Li, J. Y. Cao and X. F. Li, Electrochim. Acta, 2014, 118, 51–57 CrossRef CAS.
- H. T. T. Pham, C. Jo, J. Lee and Y. Kwon, RSC Adv., 2016, 6, 17574–17582 RSC.
- A. Hazza, D. Pletcher and R. Wills, Phys. Chem. Chem. Phys., 2004, 6, 1773–1778 RSC.
- X. H. Li, D. Pletcher and F. C. Walsh, Electrochim. Acta, 2009, 54, 4688–4695 CrossRef CAS.
- P. K. Leung, M. R. Mohamed, A. A. Shah, Q. Xu and M. B. Conde-Duran, J. Power Sources, 2015, 274, 651–658 CrossRef CAS.
- B. Li, Z. Nie, M. Vijayakumar, G. Li, J. Liu, V. Sprenkle and W. Wang, Nat. Commun., 2015, 6, 6303 CrossRef CAS PubMed.
- J. Bard and L. R. Faulkner, Electrochemical Methods-Fundamentals and Applications, Wiley, 2nd edn, 2001 Search PubMed.
- G. L. Soloveichik, Chem. Rev., 2015, 115, 11533–11558 CrossRef CAS PubMed.
- Z. P. Xie, B. Yang, D. J. Cai and L. Yang, J. Rare Earths, 2014, 32, 973–978 CrossRef CAS.
- Z. P. Xie, D. B. Zhou, F. J. Xiong, S. M. Zhang and K. L. Huang, J. Rare Earths, 2011, 29, 567–573 CrossRef CAS.
- Z. P. Xie, F. J. Xiong and D. B. Zhou, Energy Fuels, 2011, 25, 2399–2404 CrossRef CAS.
- P. K. Leung, C. Ponce-de-León, C. T. J. Low, A. A. Shah and F. C. Walsh, J. Power Sources, 2011, 196, 5174–5185 CrossRef CAS.
- L. F. Arenas, F. C. Walsh and C. Ponce-de-León, J. Electrochem. Soc., 2016, 163, A5170–A5179 CrossRef CAS.
- G. Nikiforidis, L. Berlouisa, D. Hallb and D. Hodgsonb, J. Power Sources, 2012, 206, 497–503 CrossRef CAS.
- P. Modiba, M. Matoetoe and A. M. Crouch, Electrochim. Acta, 2013, 94, 336–343 CrossRef CAS.
- Z. L. Na, S. N. Xu, D. M. Yin and L. M. Wang, J. Power Sources, 2015, 295, 28–32 CrossRef CAS.
- M. D. Gernon, M. Wu, T. Buszta and P. Janney, Green Chem., 1999, 1, 127–140 RSC.
- P. K. Leung, C. P. D. Leon, C. T. J. Low and F. C. Walsh, Electrochim. Acta, 2011, 56, 6536–6546 CrossRef CAS.
- S. Boudinar, N. Benbrahim, B. Benfedda, A. Kadri, E. Chainet and L. Hamadou, J. Electrochem. Soc., 2014, 161, D227–D234 CrossRef CAS.
- Y. T. Hsieh, Y. C. Chen and I. W. Sun, J. Electrochem. Soc., 2016, 163, D188–D193 CrossRef CAS.
- B. Li, M. Gu, Z. M. Nie, Y. Y. Shao, Q. T. Luo, X. L. Wei, X. L. Li, J. Xiao, C. M. Wang, V. Sprenkle and W. Wang, Nano Lett., 2013, 13, 1330–1335 CrossRef CAS PubMed.
- G. Wang, J. w. Chen, Y. D. Xu, B. C. Yan, X. Q. Wang, X. J. Zhu, Y. Zhang, X. J. Liu and R. L. Wang, RSC Adv., 2014, 4, 63025–63035 RSC.
- L. Z. Hu, H. J. Li, S. Y Zhu, L. S. Fan, L. H. Shi, X. Q. Liu and G. B. Xu, Chem. Commun., 2007, 40, 4146–4148 RSC.
- M. A. Pérez, O. E. L. Pérez and M. L. Teijelo, J. Electroanal. Chem., 2006, 596, 149–156 CrossRef.
- D. J. Suarez, Z. Gonzalez, C. Blanco, M. Granda, R. Menendez and R. Santamaria, ChemSusChem, 2014, 7, 914–918 CrossRef CAS PubMed.
- G. Nikifordis, L. E. A. Berlouis, D. Hall and D. Hodgson, Electrochim. Acta, 2013, 113, 412–423 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |