
Synthesis, characterization, and hole-transporting properties of pyrenyl N-substituted triazatruxenes†
Abstract
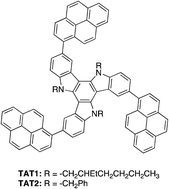
Two new pyrenyl triazatruxene derivatives (TAT1 and TAT2) were successfully synthesized via Br2-catalyzed cyclotrimerization of indole and Suzuki cross-coupling with pyrene-1-boronic acid. These compounds exhibited maximum absorption around 344–351 nm and maximum emission around 472–483 nm in CHCl3 solution. The electrochemical investigation by cyclic voltammetry (CV) suggested that the HOMO and LUMO energy levels of these compounds were around −5.3 and −2.4 eV, respectively. The performance of TAT2 as a hole-transporting material was three-times better than that of the commercial compound N,N′-bis(3-methylphenyl)-N,N′-diphenylbenzidine (TPD). The electroluminescent device of the structure ITO/PEDOT:PSS/TAT2/Alq3/LiF:Al exhibits a bright green emission with a maximum luminance of 31 971 cd m−2 at 10.8 V and a turn-on voltage of 2.6 V. In addition, the use of spin-casted films of TAT1 or TAT2 doped with 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) as greenish blue-emitting materials are demonstrated.
 Please wait while we load your content...
Please wait while we load your content...

