
Self-strengthening hybrid dental adhesive via visible-light irradiation triple polymerization†
Abstract
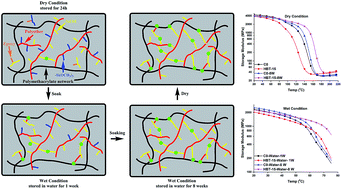
A self-strengthening methacrylate-based dental adhesive system was developed by introducing an epoxy cyclohexyl trimethoxysilane (TS) which contains both epoxy and methoxysilyl functional groups. The experimental formulation, HEMA/BisGMA/TS (22.5/27.5/50, wt%), was polymerized by visible-light. Real-time Fourier transform infrared spectroscopy (FTIR) was used to investigate in situ the free radical polymerization of methacrylate, ring-opening cationic polymerization of epoxy, and photoacid-induced sol–gel reactions. Among the three simultaneous reactions, the reaction rate of the free radical polymerization was the highest and the hydrolysis/condensation rate was the lowest. With 40 s-irradiation, the degrees of conversion of the double bond and epoxy groups at 600 s were 73.2 ± 1.2%, 87.9 ± 2.4%, respectively. Hydrolysis of the methoxysilyl group was initially <5%, and increased gradually to about 50% after 48 h dark storage. Photoacids generated through the visible-light-induced reaction were effective in catalyzing both epoxy ring-opening polymerization and methoxysilyl sol–gel reaction. The mechanical properties of copolymers made with TS concentrations from 5 to 35 wt% were obtained using dynamic mechanical analysis (DMA). In wet conditions, the storage moduli at 70 °C and glass transition temperature were significantly higher than that of the control (p < 0.05); these properties increased with TS concentration and storage time. The post reaction of hydrolysis/condensation of alkoxysilane could provide persistent strengthening whether in a neutral or acidic environment and these characteristics could lead to enhanced mechanical properties in the oral environment. The cumulative amount of leached species decreased significantly in the TS-containing copolymers. These results provide valuable information for the development of dental adhesives with reduced leaching of methacrylate monomers and enhanced mechanical properties under the wet, oral environment.
 Please wait while we load your content...
Please wait while we load your content...

