
Synthesis of quinoline acetohydrazide-hydrazone derivatives evaluated as DNA gyrase inhibitors and potent antimicrobial agents†
Abstract
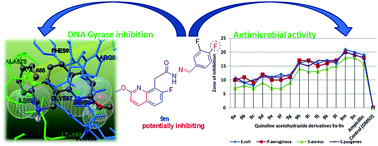
The (E)-N′-(substituted-benzylidene)-2-(7-fluoro-2-methoxyquinolin-8-yl)acetohydrazide-hydrazone derivatives reported in this manuscript represent a new series of antibacterial agents, as well as DNA gyrase inhibitors. Efforts were made to synthesize these quinolone-acetohydrazide-hydrazone derivatives (9a–n) in excellent yields. All the target compounds were evaluated for their in vitro antimicrobial activity against Escherichia coli (MTCC 443) and Pseudomonas aeruginosa (MTCC 424), as specimens of Gram-negative bacteria, and Staphylococcus aureus (MTCC 96) and Staphylococcus pyogenes (MTCC 442), as specimens of Gram-positive bacteria. Excellent antibacterial activity was observed for compounds substituted with R = 3,4,5-trimethoxy, 4-F, 4-OCF3, 4-CF3, and 3-CF3 moieties. Derivatives 9a–n were docked into the active sites of DNA gyrase A and DNA gyrase B in order to obtain more insight into the binding modes of the compounds. Among all the derivatives, 9n showed the lowest binding energy of −91.6 kcal mol−1 with DNA gyrase A. A DNA gyrase enzyme inhibition assay favors compounds 9m and 9n, which were substituted with di-fluorine moieties (R = 2,4-difluoro and 3,4-difluoro) in the quinoline scaffold, as the most potent inhibitors of S. aureus DNA gyrase A (9m, IC50 0.14 mg mL−1 and 9n, IC50 0.19 mg mL−1).
 Please wait while we load your content...
Please wait while we load your content...

