
Pretreatment and conversion of lignocellulose biomass into valuable chemicals
Jindrayani Nyoo Putroa,
Felycia Edi Soetaredjob,
Shi-Yow Lina,
Yi-Hsu Ju*a and
Suryadi Ismadji*b
aDepartment of Chemical Engineering, National Taiwan University of Science and Technology, No. 43, Sec 4, Keelung Rd., Taipei 10607, Taiwan. E-mail: yhju@mail.ntust.edu.tw
bDepartment of Chemical Engineering, Widya Mandala Surabaya Catholic University, Kalijudan 37, Surabaya 60114, Indonesia. E-mail: suryadiismadji@yahoo.com; Fax: +62 31 3891267; Tel: +62 31 3891264
First published on 4th May 2016
Abstract
In the past three decades, many studies on the production of biofuels and other chemicals have been conducted using renewable sources such as lignocellulosic biomass. Lignocellulosic biomasses are abundantly available in most countries and furthermore they are carbon neutral. However, the main problem in utilizing lignocellulosic materials lies in the recalcitrance of its bonding. This review provides a comprehensive overview and a brief discussion on producing biofuel and valuable chemicals from lignocellulose biomass. Various aspects of the physical, chemical, thermophysical, thermochemical and biological pretreatment of lignocellulosic materials are discussed in this review. The success in biofuel and chemical production strongly depends on the pretreatment method used. Overall, pretreatment is the major step in the successful production of valuable products from lignocellulosic biomass.
1. Introduction
The depleting of fossil oil resources has become a major reason to develop sustainable sources of renewable energy and chemicals.1,2 Apart from the scarcity of fossil oil as the main energy resource for transportation and industry, global warming is also considered as one of the major problems that we face today. An Intergovernmental Panel for Climate Change (IPCC) estimated that the contribution of CO2 to total greenhouse gas (GHG) is approximately 53%.3,4 In 2009, the European energy and climate package set four targets for 2020 in connection with GHG emissions: 20% reduction of GHG emissions, 20% energy efficiency improvement, 20% share of renewable energy for gross final energy usage, and 10% renewable energy in the transportation sector.5In the past decades, renewable fuels or biofuels were produced mostly from primarily food crops such as cereals, sugar cane and oil seeds (called 1st generation biofuels). Biofuels produced from these primary food crops have considerable economic value; however, their potential to meet transport fuel targets is limited by:6
• Competition for land and water used for food and fiber production,
• High production and processing costs that often require government subsidies in order to compete with petroleum products, and
• Widely varying assessments of the net GHG reductions once land-use change is taken into account.
Recently, the 2nd generation biofuels gained interests from many research groups because of the abundantly available feedstock in most countries. Lignocellulosic biomass is a renewable and carbon neutral material that can be converted into biofuel and other intermediate chemicals through various conversion routes.7 It consists of biopolymer such as cellulose (40–60%), hemicellulose (20–40%), and lignin (10–24%).8 The most common lignocellulose biomass that has been used as raw materials for chemicals derivative platform are given in Table 1. Lignocellulosic materials also have been widely utilized as intermediate liquid fuel or chemical products such as furfural, levulinic acid, and GVL.12–14
Cellulose, a crystalline polymer consists of β-linked chains, has a general formula of (C6H10O5)n. Rigidity and strength of a plant's cell wall is conferred by hydrogen bonding between the hydroxyl groups of glucose and the oxygen molecules in cellulose that creates micro fibrils which are connected in a carbohydrate matrix.15,16 Hemicellulose is a complex amorphous polymer with varying degree of branching and has lower molecular mass than cellulose. It is closely related, both chemically and structurally, to cellulose. However it differs from cellulose by the type and amount of monosaccharides that made up its structure which is generally consisted of xylose (the most abundant), galactose, glucose, arabinose, mannose and sugar acids.17 It is preferable to remove hemicellulose during pretreatment, because hemicellulose creates a cross-linked network for the structural integrity of cell walls by binding to cellulose micro-fibrils, lignin and pectin.16,18 After cellulose and hemicellulose, lignin is considered as the most abundant natural polymer on earth.19 It is the third main constituents of lignocellulosic biomass, an amorphous polymer matrix from random polymerization of three primary phenylpropane monomers: coumaryl, coniferyl and sinapyl alcohols.20,21 These three lignin precursors inflict the H (p-hydroxyphenyl), G (guaiacyl), and S (syringyl) which can be acylated and show different abundances depend on their origin.22 Since lignin is always fragmented during extraction and composes of several types of substructures which repeat in haphazard manner, it is difficult to determine the degree of its natural polymerization.23 These 3 main elements in lignocellulose material present a very complex structure and are organized together with acetyl groups, minerals and phenolic substituents.24,25 Also the utilization of lignocellulosic biomass depends on its components, because there is difference in reactivity from the interactions into extensive and complex molecular systems between cellulose, hemicellulose and lignin fractions.25 Thus, pretreatment is needed to break down the complex bonding of these 3 major components in biomass. After pretreatment, the next step is to convert them into desired chemical products. Schematic diagram of the process is shown in Fig. 1.
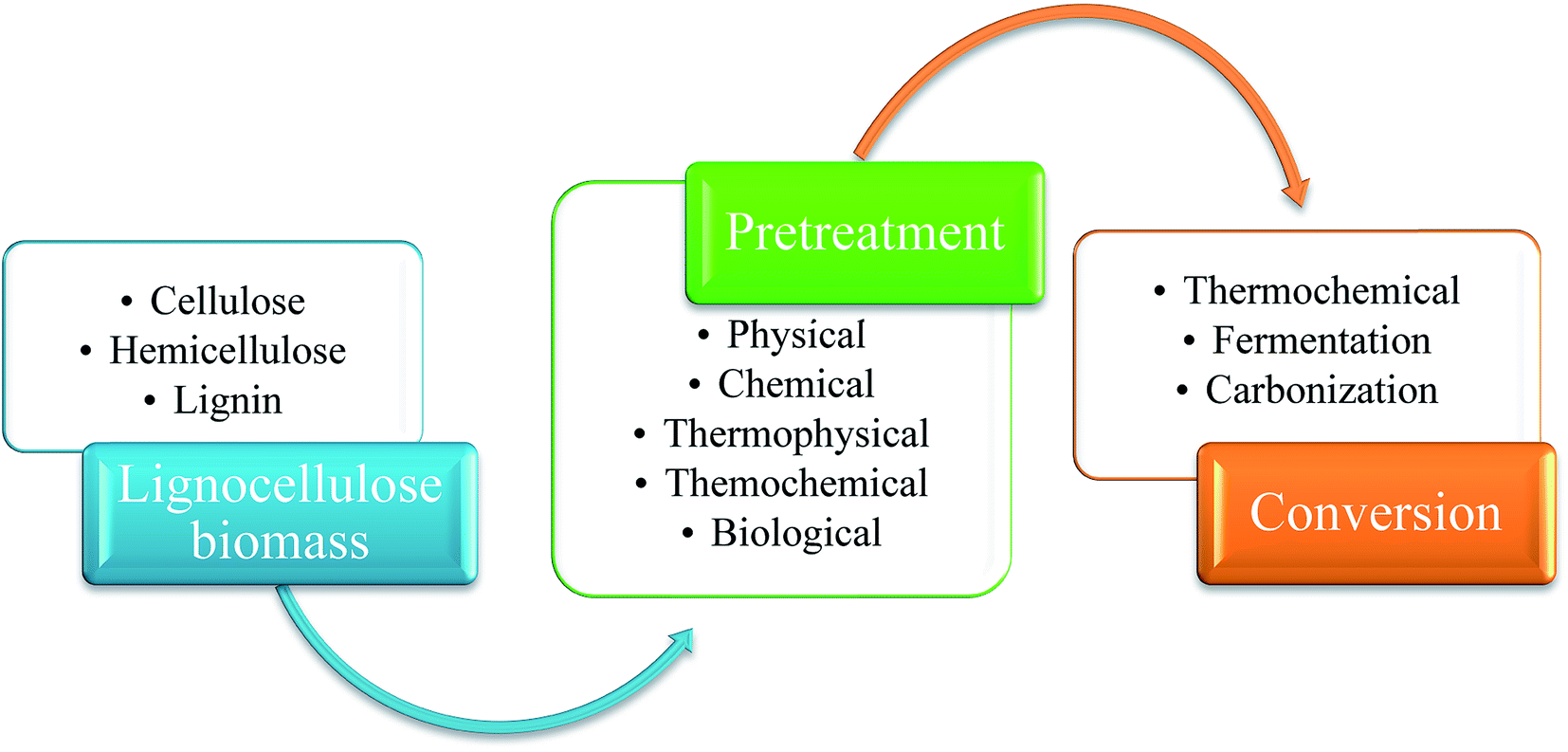 | ||
| Fig. 1 Schematic diagram showing utilization of lignocellulose biomass. | ||
Utilization of lignocellulosic biomass as raw materials for fuels and other chemicals has already been established in industrial scale, but still there is debate about the pretreatment of this material. To convert this non-edible biomass into valuable products as a sustainable source of energy and chemicals raises many challenges. One of the challenges for biofuel production is how to efficiently reduce high oxygen content from biomass and to produce biofuel with high energy density and with physical and chemical characteristics similar to fossil fuel.26 Another challenge that still need to be resolved is how to use the waste lignin after pretreatment. Lignin can be used as a feedstock to produce valuable chemicals. The focus of this review is to discuss comprehensively the pretreatment of lignocellulosic biomass, and production of high value chemical products from the pretreated biomass.
2. Pretreatment
Due to its natural recalcitrance, degradation of lignocellulosic biomass is hard. For the utilization of this material as the precursor for bio-fuel and other chemicals production, pretreatment is required to improve material accessibility. The rate of accessibility and digestibility is affected by these main factors:25,27• Crystallinity of cellulose,
• Hemicellulose disruption,
• Accessible surface area (porosity),
• Lignin protection,
• Association of cellulose–hemicellulose–lignin.
Cellulose is considered as the main contributor for the crystalline part, whereas hemicellulose and lignin are amorphous polymer. Lignin acts as a barrier to prevent cellulose and hemicellulose degradation. The removal of lignin will result in hemicellulose removal too, since lignin is chemically connected through covalent bonding with hemicellulose. Pretreatment to remove these amorphous polymers is essential to increase the specific surface area and crystallinity of cellulose.28,29
Two common types for pretreatment of this lignocellulosic material are fractionation and delignification. Fractionation is a technique to separate lignocellulosic biomass into cellulose, hemicellulose and lignin by disrupting biopolymer matrix to improve access to polysaccharides.30 The purpose of delignification is the removal of lignin, but under some conditions some hemicellulose fraction is also separated along with lignin.28,31,32 Usually delignification is included in the fractionation process to separate lignin for exposing cellulose to enzymatic hydrolysis.32 Both of these techniques actually have the same purpose. In this review, lignocellulosic biomass pretreatment will be discussed as depicted in Fig. 2.
 | ||
| Fig. 2 Various pretreatments for lignocellulosic biomass. | ||
2.1. Physical pretreatment
As is well known, crystallinity of cellulose hinders the disruption of lignocellulose material. Size reduction is a usual step to disrupt biomass crystallinity. Several studies reported the influence of distribution of biomass particle size on its conversion to biofuel.33–35 Size reduction increases the specific surface area of biomass and reduces the degree of polymerization and cellulose crystallinity; however it also depends on biomass characteristics.36,37 On the other hand, power input for mechanical size reduction depends on the moisture content of biomass, initial and final sizes. Therefore, the specific energy consumption is also affected by particle size.25,38–40 Suitable biomass particle size will obviously have impact on the design of handling, transportation and conversion facilities due to requirement of high efficiency of mass and heat transfer.41–43 Liu et al.44 studied the effect of steam explosion pretreatment on corn stover particle size for improving enzyme digestibility and reported that the amount of byproduct was higher and sugar recovery was lower for larger biomass particle size; however, sugar conversion and yield were higher during enzymatic hydrolysis. With the increase of particle size, specific surface area as well as crystallinity decrease.44 Khullar et al.45 studied the effect of particle size on enzymatic hydrolysis of pretreated Miscanthus. The highest total conversion of biomass was obtained by using the smallest particle size (0.08 mm), followed by the particle size of 2 mm, and the lowest conversion was achieved at particle size of 6 mm.45Microwave irradiation is another method of physical pretreatment of lignocellulosic biomass. This pretreatment method has been improved over many years, and is well known for its high heating efficiency and easy operation. Ma et al.46 investigated the rice straw pretreatment using microwave irradiation without the presence of any catalysts. The purpose of their study was to evaluate the influence of microwave irradiation on the recalcitrant structure, and their results showed that cellulose increased from 33.4% to 41.8%, while the acid soluble lignin decreased from 2.1% to 1.9%. This result indicates that microwave irradiation could disrupt the silicified waxy surface, break down lignin–hemicellulose complex, partially remove silicon and lignin, and expose more accessible surface area of cellulose.46
2.2. Chemical pretreatment
Utilization chemical substances to fractionate lignocellulose is widely known as pretreatment method with more advantages than physical pretreatment.38,39 During chemical pretreatment, higher glucose yield can be obtained by removing hemicellulose or lignin.47 Chemicals that are commonly used for this pretreatment48–84 are summarized in Table 2.| Chemical pretreatment | Chemicals | Effect | References |
|---|---|---|---|
| Alkaline | H2O2, NaOH, Na2SO3, Na2S, lime (CaOH2), Na2CO3, NH4OH | High lignin removal, enrichment of holocellulose, increase the porosity of biomass and cellulose swelling | 48, 50–53, 58, 61–67 and 77 |
| Acid | H2SO4, peracetic acid, HCl | Remove hemicellulose fraction and increasing biomass crystallinity | 49, 51, 52, 54–58, 62–64, 66 and 67 |
| Ionic liquids | [Bmim][OAc], [bmim][Cl], [emim][OAc], [emim][CH3COOH], [emim][DEPO4], [dmim][MeSO4], [amim][Cl], [DMSO/LiCl], [Bmpy][Cl] | Weaken the van der Walls interaction between cell wall polymers, disrupt arabinoxylan–lignin linkages, alter the fibrillar structure of cell wall, decrease cellulose crystallinity, increasing cellulose surface accessibility | 49, 59 and 68–76 |
| Organic solvent | Ethyl acetate, ethanol, acetic acid, formic acid | Break down internal lignin and hemicellulose bonds, increasing pore-volume and surface area of biomass | 60, 61 and 78–80 |
| Surfactant | Polyethylene glycol, Tween 80, Tween 20, sodium dodecyl sulfate (SDS), dodecyltrimethylammonium bromide (DoTAB), Triton X-100, Triton X-114, Agrimul NRE 1205, HM-EOPO, amphoteric Anhitole 20BS, Neopelex F-25 | Alter biomass structure, stabilizing enzyme, increasing interaction between holocellulose and enzyme, reducing adsorption of enzyme on lignin | 81–84 |
For chemical pretreatment using alkaline or acid, lignin and hemicellulose removal is affected by pH. Alkaline pretreatment using NaOH usually gives higher lignin removal than acid pretreatment using HCl and H2SO4.51,53,58,63,64 Alkaline pretreatment produces no by-product while acid pretreatment produced by-products such as 5-hydroxymethyfurfural and 2-furfuraldehyde.48,51,66 Pretreatment using alkaline hydrogen peroxide begins to gain interest due to the advantage that lignin is degraded into oxygen and water and there is no residues left in the pretreated biomass.48 In the alkali based pretreatment using NaOH, temperature only had minor impact on the lignin removal. It increased only 1% at same alkali dosage 7% w/v; but with increasing alkali dosage, the lignin removal increased from 41% to 72% at 140 °C.50 Gu et al. reported that in low temperature pretreatment, the addition of a mixture of sodium carbonate and sodium sulfate prevented the degradation of carbohydrates.65 Peracetic acid pretreatment also can remove lignin effectively and caused the degradation of some hemicellulose thus exposing cellulose.64 In addition, both acid and alkaline pretreatments removed almost all carboxylic acid substitutions such as acetyl groups and uronic acids.52 Chemical pretreatment process is widely used for industrial pulp and paper production.
Nowadays, ionic liquid (IL) is also known as one of the most promising green chemicals which can solubilize plant cell wall effectively at mild temperature.49,85 IL is called as “designer solvents” due to immeasurable cation and anion combinations,68 where the nature of cation and anion affects the solubility of biomass fraction and water interaction.76,85,86 Recently, some researchers also paid attention on the use of ILs for lignin valorization. Through catalytic oxidation of lignin, valuable platform aromatic compounds were obtained.87 Doherty et al. discussed the effect of anion composition on the efficacy of pretreatment between two ILs ([Bmim][OAc] and [Bmim][MeSO4]), their result indicated that acetate anion removed >32% of lignin from maple wood flour and significantly reduced cellulose crystallinity. As a comparison, [Bmim][MeSO4] only removed 19% of lignin without decreasing the crystallinity.88 Pretreatment using ILs also played an important role on fiber size, and the later affected the solubility of lignocellulose in solvent.70,88 Although the cost of pretreatment using ILs should be addressed carefully,76,89 process efficiency of biomass pretreatment using ILs is still better than other available conventional processes. Since IL can be recovered easily, it can overcome cost problem in industrial application.68,86,89
Another attractive chemical pretreatment is organosolv process. This pretreatment is widely known for extracting lignin from biomass using organic solvents in the presence of acidic/alkaline catalyst. This process has been used in several chemical and fuel industries.22,61,62,79,90,91 One of the advantages of this process is recovery of solvent is relatively easy, which can be conducted through various methods depending on solvent characteristics.92,93 Lignin extracted using this process had high purity and contained a small amount of phenolic and aliphatic hydroxyl.22,94,95 Without the presence of lignin, cellulose and hemicellulose fractions of the biomass can be effectively converted to platform chemicals such as 5-hydroxymethylfurfural (HMF) and levulinic acid (LA).62,96 With this organosolv pretreatment process, the major fraction in lignocellulose can be utilized as raw material for valuable platform chemicals and biofuel, and the lignin fraction could be recovered for other applications. A number of solvents with various catalysts (acid, alkaline, and chloride salt) have been used (see Table 2) to improve the fractionation process.97,98 In order to improve low recovery of hemicellulose and neutralization of acid/base, several studies reported the organosolv pretreatment of lignocellulosic materials without adding acid catalyst.94–100 The use of NaOH (1.5% NaOH for 60 min) as the catalyst resulted in higher delignification efficiency than using sulfuric acid.78 Wildschut et al.101 investigated the influence of temperature, acid and ethanol concentration on the fractionation of wheat straw, and reported that these parameters played more important roles than reaction time and particle size. Without adding any catalyst, the delignification efficiency was 37.7% while the efficiency was 75.8% with the addition of acid (30 mM of H2SO4). Xylan recovery decreased dramatically from 71.8% to 4.7% as acid concentration was increased from 0 to 30 mM.101 In the pretreatment of wheat straw, the use of organic acids gave better extraction of phenolic hydroxyl in lignin than voltaic alcohols in the degradation of hemicellulose and lignin.80,102 Organosolv process is one of the common methods for delignification of wood in the pulp and paper industries. Most common used solvents are methanol, ethanol, formic acid and acetic acid. Often these solvents are used in combination with water.
Interestingly, some articles published reported that the addition of surfactant in lignocellulose fractionation can help improving enzyme digestibility.81–84 Surfactant has amphiphilic structure that consists of hydrophilic head and hydrophobic tail. This structure of surfactant enables it to be adsorbed onto substrate thus modifies the structure of biomass.103 Surfactant can modify the surface and interfacial energy in enzymatic hydrolysis which explains the increasing rate of enzyme hydrolysis.103,104 There are five types of surfactant: non-ionic, anionic (negative charge), cationic (positive charge) and zwitterionic (positive and negative charges) and biosurfactant (produced by microorganism).103,105 Several researchers reported that non-ionic surfactant gives better result in increasing hydrolysis rate than anionic or cationic surfactant.81,84 Helle et al. observed that with the addition of surfactant, enzyme loading can be reduced. Qing et al. reported that reducing enzyme loading had greater impact on enzymatic hydrolysis.81,104 It was said that non-ionic surfactant with high value of hydrophile–lypophile balance performed better in the degradation of lignin and hemicellulose, and anionic surfactant gave poor result in hydrolysis rate.81,84,106 Surfactant in enzymatic hydrolysis was usually added at critical micelle concentration (CMC) where surfactant later formed micelle.104 If the surfactant adding was above CMC, surfactant will interact with enzyme and reducing the effectiveness of enzyme.104 The mechanism of how surfactant can increase saccharification (see Fig. 3) is that the hydrophobic part of surfactant binds with the hydrophobic part of lignin or hemicellulose and the hydrophilic part of surfactant prevents the unproductive enzyme binding with lignin, thus increases hydrolysis rate with a small amount of enzyme loading.81,84
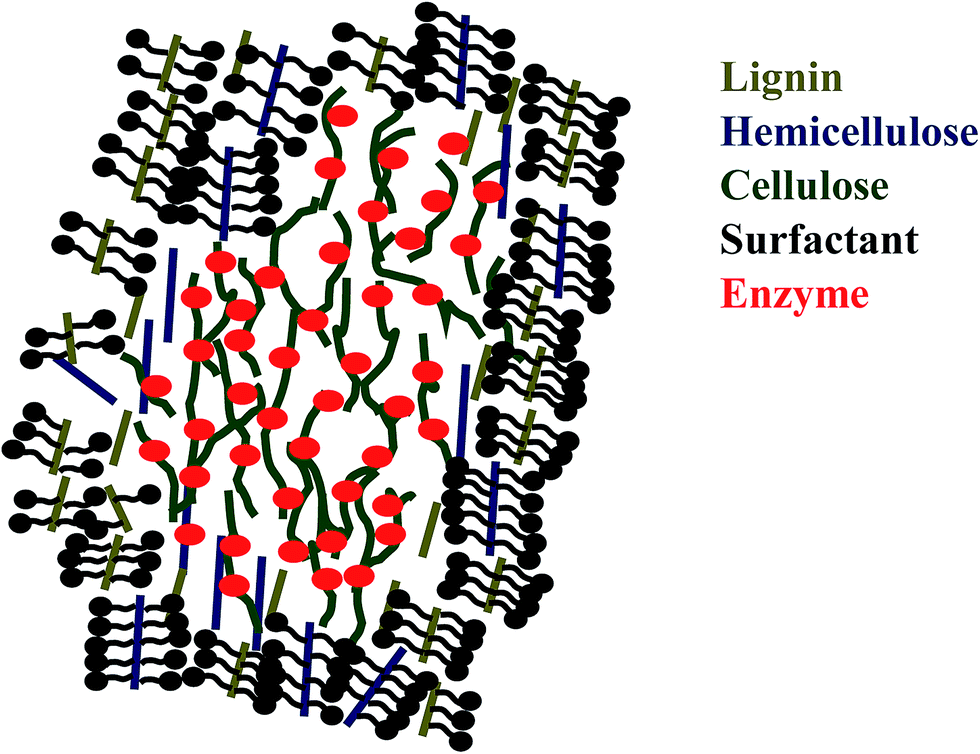 | ||
| Fig. 3 Scheme of mechanism of surfactant in saccharification. | ||
2.3. Thermo-physical and -chemical pretreatment
Considering the environmental effect, an attractive pretreatment using water as the solvent has been used for lignocellulose fractionation. Water at elevated temperature and pressure, known as liquid hot water (LHW) can be used to hydrolyze lignocellulosic biomass.107 Under high temperature and pressure, water dissociates into H3O+ and OH− ions, which can act as acid or base catalyst. Several studies reported that LHW pretreatment resulted in lower hemicellulose (mannan and xylan) content in residual biomass due to accumulation of hydrogen ion and acetyl groups in hemicellulose which can act as acids to hydrolyze hemicellulose into sugars.107,108 LHW is effective to separate xylans completely from glucans. After the separation, the major part that remains in the solid residue is glucose.109 Yu et al. compared the pretreatments of biomass using HCl, NaOH and LHW and concluded that pretreatment using HCl and LHW resulted in the same solubilization of xylan (over 86%), while pretreatment using NaOH resulted in the highest removal of lignin. Despite the high need of energy in LHW, the residue after LHW pretreatment does not need washing, which is an advantage of the process.110Steam explosion (SE) is also widely utilized for disrupting the structure of lignocellulosic materials. Generally, this pretreatment is always followed by microbial process to enhance cellulose accessibility.111–114 Some researchers also did pre-impregnation with SO2 or NaOH for better result after steam explosion; this impregnation was carried out in order to overcome non-uniformity and obtain deep penetration into biomass.115,116 The impregnation with SO2 was conducted to increase hemicellulose solubility112–114 and NaOH impregnation to increase the removal of lignin during experiment.115 Liu et al.44 discussed the effect of corn stover particle size during SE pretreatment on improving the digestibility of enzyme. Their result indicated that larger particles size improved enzymatic hydrolysis performance and gave higher pretreatment efficiency. Adapa et al.37 conducted grinding experiments on SE treated and untreated lignocellulosic materials in order to determine the effect of specific energy requirements on geometric mean particle size and distribution of lignocellulosic materials. They found that the SE pretreated biomass required less energy for grinding and particle size reduction of the untreated biomass needed considerable more energy and cost.37,44 Wiman et al.114 investigated the individual effects of pretreatment temperature, time, and sulfur dioxide uptake on cellulose accessibility. Their results concluded that cellulose accessibility increased with increasing pretreatment temperature and time. However SO2 uptake had insignificant effect on cellulose accessibility but conversion of enzymatic hydrolysis increased almost 2 times.114 This result agreed with that of Zimbardi et al. who mentioned that increasing acid loading did not show any significant improvement in water solubility but it greatly affected sugar partition between monomers and oligomers.117 Pre-impregnation using acid can cause low recovery of C5 sugar in the residue but can greatly improve enzymatic hydrolysis even though lignin content in the residue still remains high.
Pretreatment using ultrasound is considered as a promising technology in improving lignocellulosic material fractionation. In concept, ultrasound method utilizes cavitation to enhance heat and mass transfer during fractionation.118,119 Bussemaker and Zhang mentioned that oxidizing radicals were produced during ultrasonification, and these radicals played important role in the disruption of the recalcitrant lignocellulosic material.120 Several parameters in the ultrasound process such as frequency, particle size and stirring also influence the results of lignocellulosic material pretreatment (see Table 3).121 Hemicellulose sugars are bound by glycosidic linkages and are accessible to chemical and physical treatment, while lignin can be separated by chemical treatment only.120 Garcia et al. used ultrasound-assisted method for the fractionation of olive tree pruning residues using three solvents (water, aqueous acetic acid and aqueous sodium hydroxide). Their results showed that higher yield and higher selectivity were obtained by using ultrasound than that without using ultrasound. For longer ultrasound time, sodium hydroxide solution gave better separation performance than other solvents.118 The combination of ultrasound and addition of catalyst to liquefy lignocellulosic materials was studied by Kunaver et al.122 They found that the use of ultrasound in the liquefaction process inhibited the formation of large molecular structures from degradation of lignin and cellulose.
| Ultrasound pretreatment | |
|---|---|
| Frequency | • Higher frequencies can increase carbohydrate solubilization because of enhancing radical attack in consequence of increasing sonochemical effects |
| • Lower frequencies are effective for delignification due to the enhanced accessibility from the physical effects of ultrasound such as pits and cracking | |
| Particle size | • Decreasing particle size increases the carbohydrate solubilization and delignification |
| • Decreasing pH with particle size because of hemicellulose dissolution | |
| Biomass loading | •Greater delignification is achieved in the smaller solid loading of biomass |
| Stirring | • Improve fractionation of biomass (lower solid residue yield) |
| • Increase radical production at low frequencies which resulted in lower percentage of remaining lignin | |
Pretreatment of biomass in the presence of high pressure oxygen or air is called as wet oxidation. This process takes place at high temperature and effectively solubilizes hemicellulose fraction.123 Arvaniti et al. investigated the effect of temperature, time and oxygen pressure in wet oxidation of rape straw and reported that pressure played more important role than temperature and contact time on cellulose and lignin recovery. By decreasing pressure, cellulose and lignin recovery increased, while decreasing temperature and contact time gave negative effect on lignin recovery.124 Banerjee et al.125 performed wet oxidation of rice husk with addition of Na2CO3. Their result agreed with that of Schmidt and Thomsen123 in that most hemicellulose was dissolved and the solid fraction of biomass became black due to high pressure and temperature used in the process.126 The purpose of adding sodium carbonate was to adjust the pH since pH is an important factor in biomass fractionation.126 Kallioinen et al.127 investigated wet oxidation of spruce, birch, and sugar cane bagasse using different alkaline agents (NaOH, KOH or Ca(OH)2). Their result indicated that high removal of lignin was observed due to alkaline agent addition.127
One of the thermo-chemical pretreatments is the ammonia-based biomass pretreatment such as ammonia recycle percolation (ARP) and ammonia fiber/freeze explosion (AFEX). In AFEX pretreatment biomass and ammonia is enclosed in a high pressured reactor and the pressure is released rapidly to create an explosion effect. In ARP ammonia flows through biomass in the reactor and ammonia is recycled after the pretreatment.38 Due to the difference in contact of ammonia and biomass, usually ARP results in low recovery of hemicellulose and high delignification, while AFEX results in low lignin removal.38 These two processes are distinguished for their ability to enhance enzyme digestibility for the pretreated biomass which can reduce microbial need.62,128 They are also classified as alkaline pretreatment which resulted in high selectivity towards lignin, especially for ARP which can remove significant fraction of hemicellulose and lignin.38,126 The major parameters in these processes are reaction time, temperature, ammonia concentrations and loading.124 Chundawat et al.129 investigated the pretreatment of guayule using AFEX and reported that the pretreatment substantially improved overall enzyme digestibility by 4–20 folds. Kim et al.130 studied the effect of temperature and time in the ARP pretreatment of rice straw. Higher temperatures with longer reaction times increased the hydrolysis of the internal lignin and hemicellulose bonds.130 Similar result was also obtained by Bouxin et al.131 who examined the effect of ammonia concentration in the ARP pretreatment and their results indicated that decreasing ammonia concentration reduced the solubility of lignin compound of poplar sawdust. Zhao et al. studied AFEX of corn stover with and without H2O2 as the catalyst and reported that the effect of temperature and reaction time was the same as that of ARP.130,132 The addition of H2O2 in AFEX pretreatment was to increase lignin removal and sugar release.132 Ammonia loading has negligible effect on xylan and lignin removal, but glucan content increased with increasing ammonia loading.132 The increase of glucan content with ammonia loading was due to the increasing degradation of hemicellulose, removal of lignin and other soluble components.132
Supercritical CO2 (SC-CO2) is a potential thermo-physical pretreatment which is in principle similar to steam explosion. In supercritical condition, CO2 has the characteristic of a nonpolar organic solvent with low viscosity and zero surface tension which can rupture the lignocellulose structure through penetration.133 This method is usually paired with microbial attack because of vulnerable surface of biomass after SC-CO2 treatment and no inhibitory product is reduced after the treatment.134 Several parameters in the SC-CO2 treatment have been studied thoroughly, such as temperature, pressure and time. Glucose yield tends to increase with the increasing of these parameters. However, glucose yield in SC-CO2 treatment also depends on biomass characteristics.133 In SC-CO2 treatment, moisture content in biomass is an important factor because water can affect the penetration of CO2, increasing moisture content results in higher sugar yield.134,135 There are two explanations why higher moisture content gives higher sugar yield. Firstly, water and CO2 at high pressure could form carbonic acid, which increases the acid hydrolysis of hemicellulose. Second: water enables the swelling of biomass that helps CO2 penetrating deeper into the pores of biomass and disrupting biomass fibers through explosive release of pressure.133,136
2.4. Biological pretreatment
Biological pretreatment is the most expensive pretreatment method because of the high cost of certain microorganisms. Extensive studies on the use of microorganisms for pretreatment of lignocellulosic material have been conducted by various research groups, but the use of microbial for lignocellulosic material degradation is still far from industrial application. The main problem in the use of microbial process is the complex linkage of lignin–hemicellulose–cellulose, so combination with physical or chemical pretreatment is necessary before the microbial process.16,137 Initial pretreatment such as steam explosion, supercritical CO2, acid, alkaline, or organic solvent changes the physical and chemical properties of biomass which enhances enzyme digestibility. The change of biomass structure increases the digestibility of microbes due to polysaccharides modification. It should be noted that lignin removal must be carried out at low temperature to avoid sugar degradation.137,138 Some researchers reported that it is important to remove lignin for ease of enzyme attack,139 but it seems that it's not really the case. It is true that the complex linkage of lignin and carbohydrate hampers the enzyme digestibility of carbohydrate; therefore lignin needs to be removed for further conversion of lignocellulose into valuable chemical product. However some cases demonstrated that even though high lignin removal (>50%) was achieved but did not give high enzyme hydrolysis compared to low lignin removal pretreatment.113,114,140 Hence, the most important in improving enzymatic digestibility is not high lignin removal but high cellulose accessible area. With high accessible area enzyme digestibility will also greatly increase.141,142Process using microorganisms that can help removing lignin is known as biodelignification. Biodelignification can be carried out with the help of microorganism like fungi and bacteria. There are three main groups of fungus: white rot, brown rot and soft rot fungi. For bacteria there are four classes: actinomycetes, α-proteobacteria, β-proteobacteria and γ-proteobacteria.138,143 These microorganisms can degrade lignin effectively even though the conversion is slow.138,143–145 Among those microorganisms, the best one to degrade lignin effectively is white rot fungi because it exhibits highly oxidative enzymes.146 On the contrary, brown rot fungi prefer to remove carbohydrate part with partially removed lignin due to different mechanism.143 Soft rot fungi remove only soluble sugars from lignocellulose.147 White rot fungi are known to produce ligninolytic enzymes such as lignin peroxides (LiP), manganese peroxides (MnP), versatile peroxides (VP) and laccase. LiP can actively degrade phenolic and non-phenolic part of lignin, MnP and laccase can directly oxidize phenolic unit but need mediator to digest non-phenolic unit, and VP is a hybrid of LiP and MnP that can oxidize both phenolic and non-phenolic part due to dual characteristic.148,149 Brown rot fungi use Fenton oxidative reaction to generate hydroxyl radical (˙OH) and this radical will be used as an oxidant to attack lignin.143,150 Lignin degrader bacteria have individual complex pathway for specific degradation of lignin components such as β-aryl ether, biphenyl, diarylpropane, phenylcoumarane and pinoresinol.143 There are several important factors that can affect the effectiveness of fungi like fungal strain, cell wall of substrate and culture conditions.151 Saha et al. observed the behaviors of 26 white rot fungal strains on corn stover and reported that inappropriate fungal strain and biomass combination will even result in carbohydrate loss without any lignin removal.152 Except using fungi or bacteria to degrade lignin, enzyme delignification can also be considered since it offers the possibility to increase delignification efficiency and reduce process time.148 Among the ligninolytic enzymes used for delignification, laccase is widely utilized for enzyme delignification due to high removal of lignin.148 Using enzyme for delignification is easier than microorganism degradation because of wide ranges of optimum temperature and pH. The major factor is enzyme loading and solid to liquid ration in the process.148 It should be also noted that using microorganisms for biomass pretreatment produces no inhibitor, thus it will greatly facilitate the next step such as saccharification or fermentation.148
There are four different combinations between thermochemical and biological treatments which are known as separate hydrolysis and fermentation (SHF), simultaneous saccharification and fermentation (SSF), simultaneous and saccharification co-fermentation (SSCF), and consolidated bioprocessing (CBP) (see Fig. 4).39,153,154 SHF has the advantage of optimizing sacchariffication and fermentation in separated process. SSF can produce high ethanol yield with low cost. SSCF is similar to SSF but the process from saccharification until fermentation of C6 and C5 sugar occurs simultaneously, hence results in low biofuel yield. CBP has the lowest capital cost but give the lowest yield of biofuel due to the presence of inhibitors which inhibit growth of microbes. Among these four processes, the most beneficial one is SSF since it requires low initial cost and can achieve high product yield.39 IBP is a low cost process, however, the efficiency and the yield of this process are low.39 Another process is called integrated bioprocessing (IBP). Unlike previous four processes whose pretreatments are either chemical or thermophysical, in this process every step including biomass pretreatment (delignification) uses microorganism and runs in a single reactor (see Fig. 4).154,155 Therefore IBP at least needs 2 kinds of microorganism, one is for delignification and the other is for enzyme production until the fermentation step.155 It is indeed true that IBP can greatly reduce total cost and especially there is no inhibitor formation due to microbial assisted delignification which make the subsequent step easier, but until now there are no reports about lignocellulosic biomass pretreatment and process by IBP.155
 | ||
| Fig. 4 (A) Schematic process for SHF, SSF/SSCF and CBP (B) schematic process for IBP. | ||
Biological pretreatment processes are affected by parameters such as pH, temperature and inhibitor (intermediate chemical: phenolic compounds, furan derivatives, weak acids).156 The performance of several common microorganisms (Cryptococcus curvatus, Trichoderma reesei, Rhodococcus opacus, Saccharomyces cerevisiae and Kluyveromyces marxianus) for biological pretreatment in the presence of inhibitors has been studied by several research groups.157–160 The most common inhibitors present in the pretreatment of lignocellulosic biomass usually are furfural, vanillin, p-hydroxybenzaldehyde (PHB), and syringaldehyde.161 The existence of these inhibitors reduce the productivity of microorganisms.157–160 Therefore, detoxification is necessary before further step in the biological process is carried out. There are several detoxification methods such as neutralization, overliming, adsorption, ion exchange, and enzymatic detoxification which have been used effectively to remove some inhibitors (see Table 4).126 Subsequent process usually is conducted at mild temperature (20–37 °C) and pH 5–8, and these operation conditions sometimes can be a problem for scale-up in industrial application for some microorganisms.162 Several microorganisms tolerant to extreme media (low/high temperature or pH and inhibitor) have been developed during the past decades in order to improve the cost efficiency of biomass-based biofuel processes.163,164 Several microorganisms which have thermostable or thermophilic behavior have been studied to degrade lignocellulosic materials. These microorganisms offer some advantages such as shorter hydrolysis time, high resistance in low or high pH, decreasing risk of contamination and low cost of energy.165 Thermophilic bacteria also have gained much interest especially for CBP (high temperature decreases the chances of contamination) and SSF (shorter hydrolysis time which decreases potential contamination). A few examples of thermophilic bacteria that were used in the processes can be seen in Table 5.165,166
| Method | Removal of inhibitor | Note |
|---|---|---|
| Neutralization | Acetic acid, furfural and HMF | Poor ability to remove toxic compounds |
| Overliming | Furfural and HMF | Sugar loss due to hydroxide-catalyzed degradation reactions, no alter in acetic acid concentration |
| Adsorption | Furans, phenolic and acetic acid | Good removal of acetic acid and phenolic compounds |
| Ion exchange resin | Furans, phenolic and acetic acid | High removal of furan, total phenolic compounds and acetic acid |
| Electrodialysis | Acetic acid, furfural, phenolic compounds | Remove 90% of acetic acid, low sugar losses (<5%), environmental friendly, high instrument cost, better fermentability of the hydrolysate |
| Enzyme detoxification | Phenolic compounds | Excellent selectivity removal of phenolic content |
| Organism | Note |
|---|---|
| Clostridium thermocellum165 | Degrade crystalline cellulose efficiently at 60 °C and produce a large multi protein complex called cellulosome, and increase ethanol tolerance and product yields |
| Caldicellulosiruptor saccharolyticus165 | A suitable candidate for biohydrogen production, produce thermostable cellulolytic and xylanolytic enzymes, grow optimally at 70 °C on various kind of lignocellulose biomass |
| Caldicellulosiruptor bescii DSM 6725 (ref. 165 and 166) | The most thermophilic organism which grow efficiently with an optimum growth temperature of 80 °C, can degrade high concentrations of both unpretreated switchgrass and crystalline cellulose (up to 200 g L−1) |
3. Production of valuable chemical product
Many reviews have already discussed about utilizing lignocellulose biomass to produce biofuel. In this review, the authors will focus and discuss on the steps to produce valuable chemical products from the pretreated lignocellulose including biofuel, chemicals and advanced materials.3.1. Biofuel
Lignocellulose material can be converted into several kinds of biofuel such as biogas/syngas, biohydrogen, bio-oil, and bioethanol. Biogas and syngas have the same components (CO2, CH4, H2 and N2) but the process which produces them are different. Biogas is produced from the microbial assisted process and syngas is created by the partial combustion of biomass (gasification).18,167 Production of biogas is conducted by anaerobic digestion (AD), which has complex mechanism. There are four crucial steps in AD: hydrolysis, acidogenesis, acetogenesis and methanogenesis.168 Hydrolysis is always the first step in the microbial assisted process in order to break down the complex oligomers of lignocellulose.168 Acidogenesis is the fermentation step to create acidic pH while breaking down the organic matter.169 Acetogenesis is the process of acetogens that creates acetic acid, CO2 and H2O.169 The last step is methanogenesis. There are two general pathways to create methane:| CO2 + 4H2 → CH4 + 2H2O (from acidogenesis) | (1) |
| CH3COOH → CH4 + CO2 (from acetogenesis) | (2) |
Although there are two reaction mechanisms that can create methane, the main reaction is the 2nd one.169 There are at least three kinds of bacteria needed in AD, they are for acidogenesis, acetogenesis and methanogenesis.170 Syngas is produced by biomass gasification which in principle is similar to coal gasification except that biomass gasification occurs at lower temperature due to more reactive feedstock.171 In biomass gasification, basically there are three types of process: pyrolysis, partial oxidation and steam reforming.172 Pyrolysis is the thermal anaerobic decomposition of biomass at elevated temperature. Partial oxidation consumes less than the stoichiometric amount of oxygen needed, and steam gasification involves the reaction of water with biomass.171,172 Typical assumed reactions of these processes can be seen in Table 6 (based on cellulose fraction).172 Particularly biomass gasification usually involves the following steps: drying, pyrolysis, biochar gasification and combustion.173 Drying is necessary in order to reduce the moisture content of biomass. After that pyrolysis occurs for thermal breakdown of biomass. At this stage many products are produced such as tar, bio-oil and biochar that will be discussed further.173 Biochar gasification involves the following reactions between biochar and gas evolved during the process:
| Biochar + O2 → CO and CO2 |
| Biochar + CO2 → CO |
| Biochar + H2O → CH4 and CO |
| Biochar + H2 → CH4 |
| Process | Stoichiometry | Enthalphy (kJ g−1 mol−1) Tref = 1000 K |
|---|---|---|
| Pyrolysis | C6H10O5 → 5CO + 5H2 + C | 209 |
| C6H10O5 → 4CO + CH4 + C + 2H2 + H2O | −16 | |
| C6H10O5 → 3CO + CH4 + 2C + H2 + 2H2O | −152 | |
| Partial oxidation | C6H10O5 + 0.5O2 → 6CO + 5H2 | 96 |
| C6H10O5 + O2 → 5CO + CO2 + 5H2 | −180 | |
| C6H10O5 + 1.5O2 → 4CO + 2CO2 + 5H2 | −464 | |
| Steam reforming | C6H10O5 + H2O → 6CO + 6H2 | 322 |
| C6H10O5 + 3H2O → 4CO + 2CO2 + 8H2 | 276 | |
| C6H10O5 + H2O → 4CO + CO2 + CH4 + 4H2 | 85 |
Combustion is almost the same as biochar gasification, but it mainly involves O2 to create CO2 and CO as products, the reaction is exothermic.173
Pyrolysis can produce bio-oil and other products such as biochar, tar and gases. Biochar, a solid product from pyrolysis, consists mainly of carbon (∼85%).173 Tar and bio-oil, liquid product generated in the process, is an undesirable product which is formed at 200 to more than 500 °C. There are three major groups of tar composition (see Fig. 5).173 Bio-oil is produced by rapid and simultaneous depolymerization of major components in lignocellulose whose compounds generally consists of hydroxyaldehydes, hydroxyketones, sugars and dehydrosugars, carboxylic acids and phenolic compounds.173 Gases resulted from pyrolysis are divided into two groups: condensable gas (made of heavy molecular weight components, condense upon cooling) and non-condensable gas (lower molecular weight like CO2, CO, CH4, C2H6 and C2H4 that do not condense on cooling).173 Based on heating rate, pyrolysis can be classified as slow and fast pyrolysis. Although pyrolysis is an anaerobic process, sometimes it is conducted in the presence of medium such as water (hydrous pyrolysis) and hydrogen (hydro pyrolysis) to produce some chemicals. Based on vapor residence time, slow pyrolysis is divided into carbonization and conventional and fast pyrolysis is categorized as flash and ultra-rapid (Table 7).173–177 From thermal standpoint, pyrolysis can be divided into four stages: (1) drying (∼100 °C), (2) dehydration (100–300 °C), (3) primary pyrolysis (>200 °C) and (4) secondary cracking (∼300–900 °C).173 In the beginning, biomass is dried to remove free moisture.173 After that, dehydration of biomass occurs with the release of water and low molecular weight gases.173 In primary pyrolysis, most vapors or precursors of bio-oil and decomposition products of large biomass molecules (char, condensable and non-condensable gases) are produced.173 In the final stage (secondary cracking) large condensable gases with molecular weight are cracked to form additional char and gases.173
 | ||
| Fig. 5 Major groups of tar. | ||
| Based on | Pyrolysis process | Residence time | Major products |
|---|---|---|---|
| Heating rate | Slow174 | Days | Biochar |
| Fast175 | <2 s | Bio-oil | |
| Medium | Hydrous pyrolysis (H2O)176 | 45 min | Gases (CO and CO2) |
| Hydropyrolysis (H2)177 | <2 min | Bio-oil | |
| Vapor residence time173 (slow pyrolysis) | Carbonization | Days | Biochar |
| Conventional | 5–30 min | Biochar, bio-oil, gas | |
| Vapor residence time173 (fast pyrolysis) | Flash | <1 s | Bio-oil, chemicals, gas |
| Ultra-rapid | <0.5 s | Chemicals, gas |
There are five different strategies to produce bioethanol; they are SHF, SSF, SSCF, CBP and IBP as previously mentioned in Section 2.4. Among the steps in these processes, the key to produce bioethanol is fermentation. Generally, fermentation is known as the process to convert sugars into acids, alcohols or gases.178 There are two kinds of fermentation, C6 and C5 fermentation. Hexose fermentation starts with glycolysis where sugar is decomposed into pyruvate, then pyruvate is transformed by two kinds of enzyme (pyruvate decarboxylase and alcohol dehydrogenase) to produce ethanol and CO2.179,180 The reaction of hexose fermentation is depicted in Fig. 6.180 Pentose fermentation by recombinant S. cerevisiae was studied by several researchers. It is said that S. cerevisiae cannot digest xylose and arabinose but can ferment their isomer D-xylulose.180 Hence, gene encoding bacteria (xylose isomerase) or fungi (xylose reductase) which has the ability to utilize xylose and arabinose to produce D-xylulose is introduced into S. cerevisiae to improve pentose fermentation.180 Complex reaction mechanism of pentose fermentation by recombinant S. cerevisiae was well discussed by Hanh-Hägerdal.181
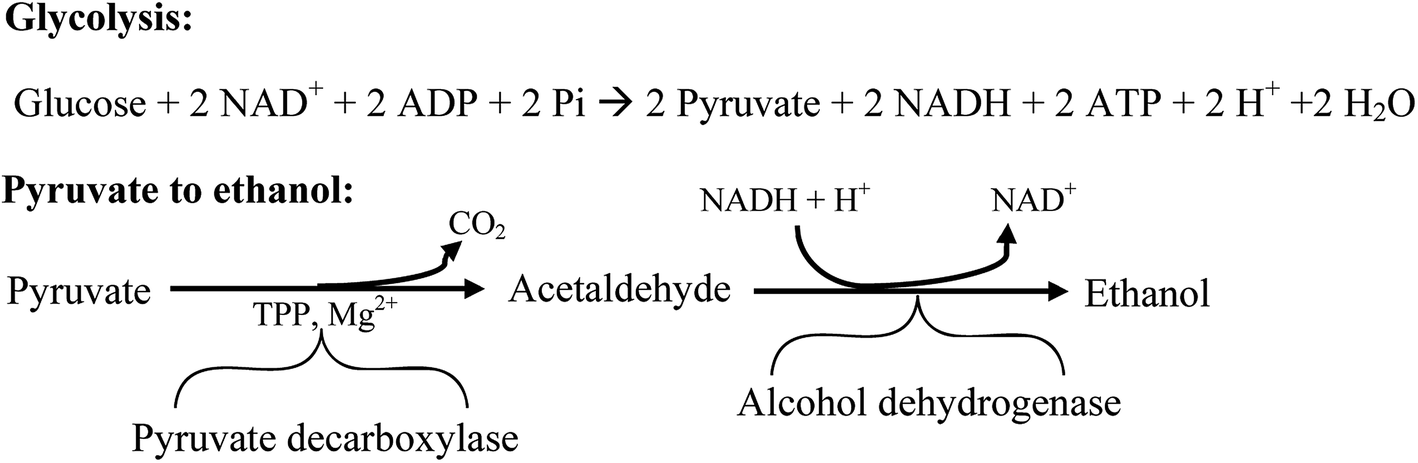 | ||
| Fig. 6 Reaction mechanism in hexose fermentation. | ||
Biohydrogen (BioH2) can be produced via thermochemical (gasification and pyrolysis) or biological routes.182 For production of H2 through pyrolysis, it can be achieved directly by fast or flash pyrolysis, while gasification can be used to produce H2 through partial oxidation and steam reformation, then further improved by water–gas shift reaction.182 The mechanism of pyrolysis and gasification can be seen in the previous paragraph. Via biological routes, there are two classifications of process using biomass as a source to produce bioH2.183 They are light dependent (photo fermentation) and light independent (dark fermentation) which have completely different mechanisms.184 Photo-fermentation uses photosynthetic bacteria which produce nitrogenase enzyme to produce H2 with the help of solar energy. The key to produce bioH2 in this process is that nitrogenase has the ability to use magnesium adenosine triphosphate and electrons to consume substrate (glucose is chosen as the precursor to represent biomass):185
| C6H12O6 + 6H2O → 24H+ + 6CO2 + 24e− → 12H2 + 6CO2 |
Dark fermentation (DF) is a process to convert biomass to bioH2 using anaerobic bacteria without light source. The common reactions during DF by facultative anaerobic microorganism are:186
| C6H12O6 + 2H2O → 2CH3COOH + 2CO2 + 4H2 | (3) |
| C6H12O6 → CH3CH2CH2COOH + 2CO2 + 2H2 | (4) |
| 4C6H12O6 + 2H2O → 3CH3CH2CH2COOH + 2CH3COOH + 8CO2 + 10H2 | (5) |
Theoretically, DF can achieve maximum yield of 4 moles H2 per mole hexose if the reaction produced only acetic acid (reaction (3)) and 2 moles H2 for butyric acid production (reaction (4)). However this situation cannot occur since the result always contains both acetic acid and butyric acid (5).182,187 Some researchers mentioned that the combination of DF and photo-fermentation can increase H2 yield since the formation of organic acid is unavoidable in DF, because photo-fermentation prefers volatile fat acids (VFA) as the substrate to sugars.188,189 According to the following reactions, a theoretical maximum yield of 12 moles H2 per mole hexose can be achieved by the combination of DF and photo-fermentation:189
| Stage 1 (DF): C6H12O6 + 2H2O → 2CH3COOH + 2CO2 + 4H2 |
| Stage 2 (photo-fermentation): 2CH3COOH + 4H2O → 8H2 + 4CO2 |
Based on the bacteria used in DF, there are three different reaction mechanisms.185 In fermentation, it always begin with glycolysis of carbohydrate to form pyruvate, after that it will be separated in three different steps to form bioH2 based on three kinds of bacteria. The first using facultative anaerobes in which pyruvate will be converted into acetyl-CoA and formate by pyruvate formate-lyase (PFL) (a) then H2 and CO2 are generated through break down of formate by formate hydrogen lyase complex (b).185 The second pathway using obligate anaerobes in which pyruvate is oxidized into acetyl-CoA and CO2 through the reduction of ferredoxin (Fd) by pyruvate ferredoxin oxidoreductase (c), then the reduced ferredoxin (Fd(red)) is re-oxidized and oxidized ferredoxin (Fd(ox)) is regenerated by [Fe–Fe] hydrogenase (HydA) together with the production of H2 (d).185 The third pathway is by thermophilic bacteria in which pyruvate formation generates NADH that reduces Fd(ox) by NADH-ferredoxin reductase (NFOR) (e), then Fd(red) generates H2 using enzyme HydA (f).185
| Pyruvate + CoA → acetyl-CoA + formate | (a) |
| Formate → H2 + CO2 | (b) |
| Pyruvate + CoA + 2Fd(ox) → acetyl-CoA + CO2 + 2Fd(red) | (c) |
| 2H+ + 2Fd(red) → H2 + 2Fd(ox) | (d) |
| 2NADH + 4Fd(ox) → 2 NAD+ + 4Fd(red) | (e) |
| 4H+ + 4Fd(red) → 2H2 + 2Fd(ox) | (f) |
3.2. Chemicals
Other chemicals such as sorbitol and xylitol can be obtained through the hydrogenation of hexose and pentose in the presence of catalyst.201–203 Glycerol is widely utilized in industry as the building block for making bio-solvents, cosmetics, batteries, polymers and surfactants.197 This substance can be produced from sugars by simultaneous hydrogenation and hydrogenolysis, or by direct hydrogenolysis of sugar alcohols (sorbitol and xylitol).204–207 Hydrogenolysis in this process is defined as hydrocracking of carbon chain that leads to the formation of shorter polyos/alcohols. Actually this process is almost similar to hydrogenation except the addition of base promoter in order to catalyze the C–C cleavage of dehydrogenation intermediate products (retro aldol derived sugars).207–210 The formation of glycerol resulted in higher yield via hydrogenolysis of sugar alcohol than sugar. In this process other glycols were also formed such as ethylene glycol (EG) and polyethylene glycol (PG).205,206
Apart from thermochemical conversion, there are two products produced by microbiology conversion: lactic and succinic acid. Microbial sources to produce lactic acid is mainly from bacteria (Bacillus sp., Streptococcus sp., Lactobacillus sp.) and mold (Rhizopus sp., Mucor sp., Monilia sp.).180,211 There are two kinds of fermentation of lactic acid, homo-fermentative and hetero-fermentative.211 In homo-fermentative, carbohydrate from lignocellulose biomass is converted by the Embden Meyerhof Parnas (EMP) glycolysis pathway that produces pyruvate and later microorganism produces lactic acid as the single product by lactate dehydrogenase.180,211 For hetero-fermentative, not only lactic acid but other minor products also appear such as ethanol, diacetyl, formate, acetoin or acetic acid and CO2.211 There are two mechanisms in hetero-fermentative: bifidus and 6P-gluconate pathway, both pathways utilize phosphoketolase enzyme to generate lactic acid from sugars with complex reactions that were discussed by Kandler.212
Unlike the previous fermentation that pyruvate is the key reactant, succinic acid is synthesized firstly through glycolysis but pyruvate will not be used to form succinic acid; it is phosphoenol pyruvate (PEP) that will be the key to form succinic acid.213–215 The reaction mechanism of producing succinic acid is very complex and depends on bacterial or fungal type used in the process. Several kinds of bacteria that have been thoroughly studied are Actinobacillus succinogenes, Anaerobiospirillum succiniciproducens, Mannheimia succiniciproducens and recombinant Escherichia coli.180,213 All these bacteria form mixed acid fermentation that produces a mixture of products including succinic acid, ethanol, lactic acid, formic acid and acetic acid; from which succinic acid must be separated.180 For A. succinogenes and A. succiniciproducens, they form succinate acid via PEP carboxykinase pathway using four key enzymes: PEP carboxykinase, malate dehydrogenase, fumarase and fumarate dehydrogenase.214 In M. succiniciproducens there are seven key enzymes (PEP carboxykinase/carboxylase, pyruvate kinase, oxaloacetate decarboxylase, malate dehydrogenase, malic enzyme, fumarase and fumarate reductase) to produce succinate.213 While recombinant E. coli has six different pathways with PEP carboxykinase plays the minor role, which causes lower yield of succinic acid production.213 Interestingly, the PEP carboxykinase pathway in bacteria fermentation is adjusted by CO2 level where higher CO2 level will produce higher yield of succinic acid.213,214 The industry application of succinic acid is huge, especially in these four markets: surfactant/detergent extender/foaming agent, ion chelator, food market (acidulant/pH modifier, flavoring agent, anti-microbial agent) and health related agent.214 The development of these chemical building blocks is very advanced now due to dwindling supply of petroleum oil and climate change problem that haunted future generations on earth.
 | ||
| Fig. 7 Overall reaction mechanisms of lignocellulose to chemicals. F: fermentation, Hl: hydrolysis, dH: dehydration, rH: rehydration, O: oxidation, H: hydrogenation, Hg: hydrogenolysis, dP: depolymerization. | ||
3.3. Advanced material
Apart from bio-fuels and chemicals, lignocellulose biomass can also be utilized for environmental remediation and development of advanced materials such as adsorbent, nanocomposites; for energy storage, transportation, medical application in drug delivery and biosensing.226–230 A few works reported using lignocellulose-based material as biosorbent for heavy metal or dye removal.227,231 Adsorption using biosorbent is not restricted by physical bonding, it may involve strong interaction between sorbent and solute molecules.232 Lignocellulose biomass can be utilized directly for adsorption or chemically modified (commonly using base or acid) to enhance adsorption capacity.231 In general, chemically modified biosorbent has better adsorption performance due to formation of new functional group that creates more active binding sites.231Electric double layer capacitor (EDLC) or supercapacitor is an energy storage device. It uses carbon as the active material which can be derived from lignocellulose biomass.226 Supercapacitor from lignocellulose can be created by the hydrothermal carbonization (HTC) process which is classified into high temperature and low temperature HTC.233 High temperature HTC (>300 °C) usually produces carbon nanotubes, graphite and activated carbon materials, while low temperature HTC (<300 °C) produces various carbonaceous materials with different sizes and shapes.233
Several works reported producing supercapacitors from lignocellulose biomass such as cornstalk, spruce, corncob, cassava peel, water hyacinth.226,234–237 Wang et al. converted cornstalk into porous graphitic carbon nanosheets by pyrolysis at high temperature (1000 to 1200 °C).226 Kurniawan et al. produced carbon microsphere from water hyacinth using subcritical water instead of pyrolysis, which is known as an environmental friendly method.236 Several reaction mechanisms can occur in the HTC process such as hydrolysis, dehydration, decarboxylation, polymerization and aromatization.238 These reactions did not occur consecutively, but appeared as a parallel network of different reaction paths that primarily depend on the type of feed.238
Besides supercapacitors, HTC process also produces another product called carbon fiber. Conventionally the precursor used in carbon fiber production is lignin isolated from lignocellulose biomass. Usually lignin obtained from lignocellulose biomass pretreatment is purified, then processed using several processes such as spinning, thermostabilization and carbonization to generate carbon fiber.239 Soenjaya et al. produced carbon fiber from water hyacinth through pyrolysis. Tar from pyrolysis was extracted to obtain phenolic compounds. These phenolic compounds then were utilized as the raw material for producing carbon fiber.240
Cellulose is the most abundant renewable polymer in the world. For hundreds of centuries it has been used as sources for energy, textile and building materials.228 This natural polymer can be used as the raw material for producing nanoscale material known as nanocrystalline cellulose (NCC). NCC has a diameter of 5–70 nm and a length between 100 and 250 nm. It exhibits extraordinary properties such as high tensile strength (7500 MPa), high rigidity (100–140 GPa) and large surface area (150–250 m2 g−1).230 Due to these remarkable properties, NCC is considered as one of the strongest and stiffest materials in the world.230 Cellulose is also utilized as the raw material for cellulose nanofibrils (CNF) and cellulose microfibrils (CMF) production.241 Both of these materials contain amorphous and crystalline parts of cellulose.228 CNF has nanoscale diameter like NCC, but microscale length.242 There are several ways to obtain CNF, using either mechanical or chemical treatment. For mechanical treatment, techniques commonly used are homogenization, cryocrushing, grinding and microfluidization. The main purpose of these processes is to defibrillate fibers.243,244 Combination of enzymatic or chemical hydrolysis with mechanical treatment also has been used in order to reduce high energy consumption of mechanical treatment.245,246 Ultrasonication also has been used to isolate CNF. This process uses acoustic cavitation to induce microjets and shock waves on microfibers. Thus, it can break the van der Waals molecular interactions among nanofibers.247
Generally, there are two steps to produce NCC from cellulose fiber: hydrolysis of the amorphous region of cellulose fiber and fragmentation of crystalline part to produce NCC.248 Acid hydrolysis is employed to remove amorphous part of cellulose. Sulfuric acid is commonly used for acid hydrolysis under strictly controlled conditions of temperature, agitation, time and acid to cellulose ratio.249 The types of acid used is very important in NCC preparations. Besides sulfuric acid hydrochloric acid also has been used to hydrolyze cellulose fiber, but resulted in flocculating aqueous suspensions.228,249 In contrast, sulfuric acid as hydrolyzing agent introduces sulfate ions onto hydroxyl groups that prevents aqueous suspensions from agglomeration.250 Normally, rod-like nanocrystal morphology were obtained by using either hydrochloric or sulfuric acid.249 Combining sulfuric and hydrochloric acid under sonication will give spherical NCC with better thermal stability than rod-like shaped NCC.
Oxidation using ammonium persulfate is believed to give more homogeneous NCC than acid hydrolysis.230 Post treatment like mechanical or sonication is conducted after acid hydrolysis in order to disperse nanocrystals into a stable suspension.228 Drying is an important step in NCC/NCF preparation. Due to the hydrophilic nature of cellulose, hydrogen bonds of cellulose tend to aggregate forming bulky material that spoils the nanostructure material.228,251 Therefore, other drying methods usually considered are freeze-drying, supercritical drying or spray drying to keep the nanoscale dimension of CNF or NCC.252
4. Concluding remarks
The main purpose of using lignocellulosic biomass as raw material for biofuels and chemicals production is that it is renewable and environmental friendly. Success of the process strongly depends on the pretreatment method used to remove lignin from cellulose or hemicellulose. To the present, various methods are available for the pretreatment of lignocellulosic material and these pretreatment methods are categorized as physical, chemical, thermophysical, thermochemical and biological pretreatment.Chemical pretreatments require chemical substances to extract lignin from the structure of lignocellulosic material, and most chemicals used will end up as waste which need further treatment prior to its release to environment. Thermophysical and thermochemical pretreatments are mostly conducted at high temperature and high pressure, and often the addition of chemical substances as catalyst is needed. In terms of cost and complexity of process, thermophysical and thermochemical pretreatment processes are expensive. Since thermophysical and thermochemical pretreatments are operated at pressure between 10 and 50 bar and temperature between 100 and 250 °C, special design and material of construction for delignification reactor are required. These become the main obstacle for biofuels and chemicals production in large scale.
Although development in biological treatment of lignocellulosic material has improved considerably, some considerations are still needed before it can be implemented in industrial scale. Factors such as oxygen supply (low gas solubility at elevated temperature), existence of inhibitors, energy consumption, economy value, waste production and growth of microorganism need to be considered. Deep and comprehensive studies are still required in order to make the biological pretreatment viable for industrial scale in terms of efficiency of energy and cost.
References
- A. Llamas, A. M. Al-lal, M. Hernandez, M. Lapuerta and L. Canoira, Energy Fuels, 2012, 26, 5968–5976 CrossRef CAS.
- G. Liu, B. Yan and G. Chen, Renewable Sustainable Energy Rev., 2003, 25, 59–70 CrossRef.
- S. Sgouridis, P. A. Bonnefoy and R. J. Hansman, Transport. Res. Pol. Pract., 2011, 45, 1077–1091 CrossRef.
- E. Corporan, T. Edwards, L. Shafer, M. J. DeWitt, C. Klingshirn, S. Zabarnick, Z. West, R. Striebich, J. Graham and J. Klein, Energy Fuels, 2011, 25, 955–966 CrossRef CAS.
- H. Schwaiger, A. Tuerk, N. Pena, J. Sijm, A. Arrasto and C. Kettner, Biomass Bioenergy, 2012, 38, 102–108 CrossRef.
- R. E. H. Sims, W. Mabee, J. N. Saddler and M. Taylor, Bioresour. Technol., 2010, 101, 1570–1580 CrossRef CAS PubMed.
- S. Liu, L. P. Abrahamson and G. M. Scott, Biomass Bioenergy, 2012, 39, 1–4 CrossRef.
- P. Daorattanachai, S. Namuangruk, N. Viriya-empikul, N. Laosiripojana and K. Faungnawakij, J. Ind. Eng. Chem., 2012, 18, 1893–1901 CrossRef CAS.
- J. K. Kurian, G. R. Nair, A. Hussain and G. S. V. Raghavan, Renewable Sustainable Energy Rev., 2013, 25, 205–219 CrossRef.
- V. Menon and M. Rao, Prog. Energy Combust. Sci., 2012, 38, 522–550 CrossRef CAS.
- S. M. Sen, C. A. Henao, D. J. Braden, J. A. Dumesic and C. T. Maravelias, Chem. Eng. Sci., 2012, 67, 57–67 CrossRef.
- L. Zhang, H. Yu, P. Wang, H. Dong and X. Peng, Bioresour. Technol., 2013, 130, 110–116 CrossRef CAS PubMed.
- B. Girisuta, K. Dussan, D. Haverty, J. J. Leahy and M. H. B. Hayes, Chem. Eng. J., 2013, 217, 61–70 CrossRef CAS.
- X. L. Du, Q. Y. Bi, Y. M. Liu, Y. Cao and K. N. Fan, ChemSusChem, 2011, 4, 1838–1843 CrossRef CAS PubMed.
- E. G. Rodriguez, O. M. P. Rivera, L. J. Enriquez, J. A. Ramirez and M. Vazquez, Biomass Bioenergy, 2012, 36, 346–355 CrossRef.
- Z. Zhang, A. A. Donaldson and X. Ma, Biotechnol. Adv., 2012, 30, 913–919 CrossRef CAS PubMed.
- P. Strunk, PhD thesis, Umea University, 2012.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- Y. H. Ju, L. H. Huynh, N. S. Kasim, T. J. Guo, J. H. Wang and A. E. Fazary, Carbohydr. Polym., 2011, 83, 591–599 CrossRef CAS.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84(A5), 339–349 CrossRef CAS.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- R. E. Hage, N. Brosse, L. Chrusciel, C. Sanchez, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2009, 94, 1632–1638 CrossRef.
- W. O. S. Doherty, P. Mousavioun and C. M. Fellows, Ind. Crops Prod., 2011, 33, 259–276 CrossRef CAS.
- F. L. Digabel and L. Averous, Carbohydr. Polym., 2006, 66, 537–545 CrossRef.
- A. Barakat, H. D. Vries and X. Rouau, Bioresour. Technol., 2013, 134, 362–373 CrossRef CAS PubMed.
- J. A. Melero, J. Iglesias and A. Garcia, Energy Environ. Sci., 2012, 5, 7393–7420 CAS.
- S. I. Njoku, B. K. Ahring and H. Uellendahl, Bioresour. Technol., 2012, 124, 105–110 CrossRef CAS PubMed.
- Y. P. Timilsena, C. J. Abeywickrama, S. K. Rakshit and N. Brosse, Bioresour. Technol., 2013, 135, 82–88 CrossRef CAS PubMed.
- F. Xu, Y. C. Shi and D. Wang, Carbohydr. Polym., 2013, 94, 904–917 CrossRef CAS PubMed.
- J. Y. Lee, H. J. Ryu and K. K. Oh, Bioresour. Technol., 2013, 132, 84–90 CrossRef CAS PubMed.
- X. Duan, C. Zhang, X. Ju, Q. Li, S. Chen, J. Wang and Z. Liu, Bioresour. Technol., 2013, 140, 363–367 CrossRef CAS PubMed.
- X. Ju, M. Engelhard and X. Zhang, Bioresour. Technol., 2013, 132, 137–145 CrossRef CAS PubMed.
- C. R. Cardoso, T. J. P. Oliveira, J. A. S. Junior and C. H. Ataide, Powder Technol., 2013, 245, 105–114 CrossRef CAS.
- C. Drieimeier, M. M. Oliveira, F. M. Mendes and E. O. Gomes, Powder Technol., 2011, 214, 111–116 CrossRef.
- Q. Zhang, P. Zhang, Z. J. Pei and D. Wang, Renewable Energy, 2013, 60, 127–136 CrossRef CAS.
- N. Saadaoui, A. Rouilly, K. Fares and L. Rigal, Mater. Des., 2013, 50, 302–308 CrossRef CAS.
- P. Adapa, L. Tabil and G. Schoenau, Biomass Bioenergy, 2011, 35, 549–561 CrossRef CAS.
- V. B. Agbor, N. Cicek, R. Sparling, A. Berlin and D. B. Levin, Biotechnol. Adv., 2011, 29, 675–685 CrossRef CAS PubMed.
- N. Sarkar, S. K. Ghosh, S. Bannerjee and K. Aikat, Renewable Energy, 2012, 37, 19–27 CrossRef CAS.
- M. N. Islam and R. Matzen, Powder Technol., 1988, 54, 235–241 CrossRef.
- V. S. P. Bitra, A. R. Womac, Y. T. Yang, C. Igathinathane, P. I. Miu, N. Chevanan and N. Sokhansanj, Bioresour. Technol., 2009, 100, 5176–5188 CrossRef CAS.
- G. G. D. Silva, S. Guilbert and X. Rouau, Powder Technol., 2011, 208, 266–270 CrossRef CAS.
- K. Y. Chiang, K. L. Chien and C. H. Lu, Appl. Energy, 2012, 100, 164–171 CrossRef CAS.
- Z. H. Liu, L. Qin, F. Pang, M. J. Jin, B. Z. Li, Y. Kang, B. E. Dale and Y. J. Yuan, Ind. Crops Prod., 2013, 44, 176–184 CrossRef CAS.
- E. Khullar, B. S. Dien, K. D. Rausch, M. E. Tumbleson and V. Singh, Ind. Crops Prod., 2013, 44, 11–17 CrossRef CAS.
- H. Ma, W. W. Liu, X. Chen, Y. J. Wu and Z. L. Yu, Bioresour. Technol., 2009, 100, 1279–1284 CrossRef CAS PubMed.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS PubMed.
- J. A. D. C. Correia, J. E. M. Junior, L. R. B. Goncalves and M. V. P. Rocha, Bioresour. Technol., 2013, 139, 249–256 CrossRef PubMed.
- C. Li, B. Knierim, C. Manisseri, R. Arora, H. V. Scheller, M. Auer, K. P. Vogel, B. A. Simmons and S. Singh, Bioresour. Technol., 2010, 101, 4900–4906 CrossRef CAS.
- Q. Li, Y. Gao, H. Wang, B. Li, C. Liu, G. Yu and X. Mu, Bioresour. Technol., 2012, 125, 193–199 CrossRef CAS PubMed.
- R. J. Garlock, V. Balan, B. E. Dale, V. R. Pallapolu, Y. Y. Lee, Y. Kim, N. S. Mosier, M. R. Ladisch, M. T. Holtzapple, M. Falls, R. S. Ramirez, J. Shi, M. A. Ebrik, T. Redmont, B. Yang, C. E. Wyman, B. S. Donohoe, T. B. Vinzant, R. T. Elander, B. Hames, S. Thomas and R. E. Warner, Bioresour. Technol., 2011, 102, 11063–11071 CrossRef CAS PubMed.
- C. E. Wyman, V. Balan, B. E. Dale, R. T. Elander, M. Falls, B. Hames, M. T. Holtzapple, M. R. Ladisch, Y. Y. Lee, N. Mosier, V. R. Pallapolu, J. Shi, S. R. Thomas and R. E. Warner, Bioresour. Technol., 2011, 102, 11052–11062 CrossRef CAS PubMed.
- Y. C. Park and J. S. Kim, Energy, 2012, 47, 31–35 CrossRef CAS.
- Y. Sun and J. J. Cheng, Bioresour. Technol., 2005, 96, 1599–1606 CrossRef CAS PubMed.
- C. Martin, B. Alriksson, A. Sjode, N. O. Nilvebrant and L. J. Jonsson, Appl. Biochem. Biotechnol., 2007, 136–140, 339–352 CrossRef PubMed.
- B. S. Baboukani, M. Vossoughi and I. Alemzadeh, Biosystems Eng., 2012, 111, 166–174 CrossRef.
- I. A. Panagiotopoulos, R. R. Bakker, T. D. Vrije and E. G. Koukios, Bioresour. Technol., 2011, 102, 11204–11211 CrossRef CAS PubMed.
- R. A. Silverstein, Y. Chen, R. R. S. Shivappa, M. D. Boyette and J. Osborne, Bioresour. Technol., 2007, 98, 3000–3011 CrossRef CAS PubMed.
- N. Labbe, L. M. Kline, L. Moens, K. Kim, P. C. Kim and D. G. Hayes, Bioresour. Technol., 2012, 104, 701–707 CrossRef CAS PubMed.
- I. Cybulska, G. P. Brudecki, B. R. Hankerson, J. L. Julson and H. Lei, Bioresour. Technol., 2013, 127, 92–99 CrossRef CAS PubMed.
- K. Wormeyer, T. Ingram, B. Saake, G. Brunner and I. Smirnova, Bioresour. Technol., 2011, 102, 4157–4164 CrossRef PubMed.
- L. D. C. Sousa, S. P. S. Chundawat, V. Balan and B. E. Dale, Curr. Opin. Biotechnol., 2009, 20, 339–347 CrossRef CAS PubMed.
- K. C. Nlewem and M. E. Trash Jr, Bioresour. Technol., 2010, 101, 5426–5430 CrossRef CAS PubMed.
- X. Zhao, L. Zhang and D. Liu, Bioresour. Technol., 2008, 99, 3729–3736 CrossRef CAS PubMed.
- F. Gu, L. Yang, Y. Jin, Q. Han, H. M. Chang, H. Jameel and R. Phillips, Bioresour. Technol., 2012, 124, 299–305 CrossRef CAS PubMed.
- M. Pedersen, A. V. Nielsen and A. S. Meyer, Process Biochem., 2010, 45, 1181–1186 CrossRef CAS.
- R. Gupta, Y. P. Khasa and R. C. Kuhad, Carbohydr. Polym., 2011, 84, 1103–1109 CrossRef CAS.
- A. M. D. C. Lopes, K. G. Joao, D. F. Rubik, E. B. Lukasik, L. C. Duarte, J. Andreaus and R. B. Lukasik, Bioresour. Technol., 2013, 142, 198–208 CrossRef PubMed.
- P. Weerachanchai, S. S. J. Leong, M. W. Chang, C. B. Ching and J. M. Lee, Bioresour. Technol., 2012, 111, 453–459 CrossRef CAS PubMed.
- T. Leskinen, A. W. T. King, I. Kilpelainen and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2011, 50, 12349–12357 CrossRef CAS.
- T. Leskinen, A. W. T. King, I. Kilpelainen and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2013, 52, 3958–3966 CrossRef CAS.
- L. Y. Meng, S. M. Kang, X. M. Zhang, Y. Y. Wu, F. Xu and R. C. Sun, Bioresour. Technol., 2012, 110, 308–313 CrossRef CAS PubMed.
- S. Singh, P. Varanasi, P. Singh, P. D. Adams, M. Auer and B. A. Simmons, Biomass Bioenergy, 2013, 54, 276–283 CrossRef CAS.
- J. A. P. Pimienta, M. G. L. Ortega, P. Varanasi, V. Stavila, G. Cheng, S. Singh and B. A. Simmons, Bioresour. Technol., 2013, 127, 18–24 CrossRef PubMed.
- H. T. Tan and K. T. Lee, Chem. Eng. J., 2012, 183, 448–458 CrossRef CAS.
- Uju, Y. Shoda, A. Nakamoto, M. Goto, W. Tokuhara, Y. Noritake, S. Katahira, N. Ishida, K. Nakashima, C. Ogino and N. Kamiya, Bioresour. Technol., 2012, 103, 446–452 CrossRef CAS PubMed.
- S. Kim and M. T. Holtzapple, Bioresour. Technol., 2005, 96, 1994–2006 CrossRef CAS PubMed.
- L. Mesa, E. Gonzales, E. Ruiz, I. Romero, C. Cara, F. Felissia and E. Castro, Appl. Energy, 2010, 87, 109–114 CrossRef CAS.
- B. W. Koo, B. C. Min, K. S. Gwak, S. M. Lee, J. W. Choi, H. Yeo and I. G. Choi, Biomass Bioenergy, 2012, 42, 24–32 CrossRef CAS.
- F. Xu, C. F. Liu, Z. C. Geng, J. X. Sun, R. C. Sun, B. H. Hei, L. Lin, S. B. Wu and J. Je, Polym. Degrad. Stab., 2006, 91, 1880–1886 CrossRef CAS.
- Q. Qing, B. Yang and C. E. Wyman, Bioresour. Technol., 2010, 101, 5941–5951 CrossRef CAS PubMed.
- H. Ooshima, M. Sakata and Y. Harano, Biotechnol. Bioeng., 1986, 28, 1727–1734 CrossRef CAS PubMed.
- M. Castanon and C. R. Wilke, Biotechnol. Bioeng., 1980, 22, 1037–1053 CrossRef CAS.
- T. Eriksson, J. Börjesson and F. Tjerneld, Enzyme Microb. Technol., 2002, 31, 353–364 CrossRef CAS.
- T. Vancov, A. Alston, T. Brown and S. McIntosh, Renewable Energy, 2012, 45, 1–6 CrossRef CAS.
- C.-Z. Liu, F. Wang, A. R. Stiles and C. Guo, Appl. Energy, 2012, 92, 406–414 CrossRef CAS.
- G. Chatel and R. D. Rogers, ACS Sustainable Chem. Eng., 2013, 2, 322–339 CrossRef.
- T. V. Doherty, M. Mora-Pale, S. E. Foley, R. J. Lindhardt and J. S. Dordick, Green Chem., 2010, 12, 1967–1975 RSC.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- A. Garcia, M. G. Alriols and J. Labidi, Ind. Crops Prod., 2014, 53, 102–110 CrossRef CAS.
- B. W. Koo, H. Y. Kim, N. Park, S. M. Lee, H. Yeo and I. G. Choi, Biomass Bioenergy, 2011, 35, 1833–1840 CrossRef CAS.
- L. P. Cantu, A. Schreiber, F. Schutt, B. Saake, C. Kirsch and I. Smirnova, Bioresour. Technol., 2013, 142, 428–435 CrossRef PubMed.
- J. Viell, A. Harwardt, J. Seiler and W. Marquardt, Bioresour. Technol., 2013, 150, 89–97 CrossRef CAS PubMed.
- J. Snelders, E. Dornez, B. B. Mlayah, W. J. J. Huijgen, P. J. de Wild, R. J. A. Gosselink, J. Gerritsma and C. M. Courtin, Bioresour. Technol., 2014, 156, 275–282 CrossRef CAS PubMed.
- A. Vishtal and A. Kraslawski, BioResources, 2011, 6(3), 3547–3568 Search PubMed.
- P. Azadi, R. C. Flores, Y. J. P. Torres, E. I. Gurbuz, R. Farnood and J. A. Dumesic, Green Chem., 2012, 14, 1573–1576 RSC.
- M. J. de la Torre, A. Moral, M. D. Hernandez, E. Cabeza and A. Tijero, Ind. Crops Prod., 2013, 45, 58–63 CrossRef CAS.
- Z. M. A. Bundhoo, A. Mudhoo and R. Mohee, Crit. Rev. Environ. Sci. Technol., 2013, 43, 2140–2211 CrossRef CAS.
- L. Kupiainen, J. Ahola and J. Tanskanen, Bioresour. Technol., 2012, 116, 29–35 CrossRef CAS PubMed.
- A. Geng, F. Xin and J. Y. Ip, Bioresour. Technol., 2012, 104, 715–721 CrossRef CAS PubMed.
- J. Wildschut, A. T. Smit, J. H. Reith and W. J. J. Huijgen, Bioresour. Technol., 2013, 135, 58–66 CrossRef CAS PubMed.
- X. Erdocia, R. Prado, M. A. Corcuera and J. Labidi, J. Ind. Eng. Chem., 2014, 20, 1103–1108 CrossRef CAS.
- D. Myers, Surfaces, Interfaces, and Colloids: Principles and Applications, John Wiley & Sons, New York, 2nd edn, 1991, pp. 21–22 Search PubMed.
- S. S. Helle, S. J. B. Duff and D. G. Cooper, Biotechnol. Bioeng., 1993, 42, 611–617 CrossRef CAS PubMed.
- C. N. Mulligan, Environ. Pollut., 2005, 133(2), 183–198 CrossRef CAS PubMed.
- M. Kurakake, H. Ooshima, J. Kato and Y. Harano, Bioresour. Technol., 1994, 49, 247–251 CrossRef CAS.
- C. Wan, Y. Zhou and Y. Li, Bioresour. Technol., 2011, 102, 6254–6259 CrossRef CAS PubMed.
- C. Wan and Y. Li, Bioresour. Technol., 2011, 102, 9788–9793 CrossRef CAS PubMed.
- T. Ingram, T. Rogalinski, V. Bockemuhl, G. Antrakinian and G. Brunner, J. Supercrit. Fluids, 2009, 48, 238–246 CrossRef CAS.
- Q. Yu, X. Zhang, S. Lv, M. He, Y. Zhang, Z. Yuan, W. Qi, Q. Wang, W. Wang and X. Tan, Bioresour. Technol., 2013, 129, 592–598 CrossRef CAS PubMed.
- K. Ohgren, R. Bura, G. Lesnicki, J. Saddler and G. Zacchi, Process Biochem., 2007, 42, 834–839 CrossRef.
- C. Tengborg, M. Galbe and G. Zacchi, Enzyme Microb. Technol., 2001, 28, 835–844 CrossRef CAS PubMed.
- S. Nakagame, R. P. Chandra, J. F. Kadla and J. N. Saddler, Bioresour. Technol., 2011, 102, 4507–4517 CrossRef CAS PubMed.
- M. Wiman, D. Dienes, M. A. T. Hansen, T. V. D. Meulen, G. Zacchi and G. Liden, Bioresour. Technol., 2012, 126, 208–215 CrossRef CAS PubMed.
- K. M. F. Kazi, P. Jollez and E. Chornet, Biomass Bioenergy, 1998, 15(2), 125–141 CrossRef CAS.
- S. Monavari, M. Galbe and G. Zacchi, Bioresour. Technol., 2009, 100, 6312–6316 CrossRef CAS PubMed.
- F. Zimbardi, E. Viola, F. Nanna, E. Larocca, M. Cardinale and D. Barisano, Ind. Crops Prod., 2007, 26, 195–206 CrossRef CAS.
- A. Garcia, M. G. Alriols, R. L. Ponte and J. Labidi, Bioresour. Technol., 2011, 102, 6326–6330 CrossRef CAS PubMed.
- J. Luo, Z. Fang and R. L. Smith Jr, Prog. Energy Combust. Sci., 2014, 41, 56–93 CrossRef.
- M. J. Bussemaker and D. Zhang, Ind. Eng. Chem. Res., 2013, 52, 3563–3580 CrossRef CAS.
- M. J. Bussemaker, F. Xu and D. Zhang, Bioresour. Technol., 2013, 148, 15–23 CrossRef CAS PubMed.
- M. Kunaver, E. Jasiukaityte and N. Cuk, Bioresour. Technol., 2012, 103, 360–366 CrossRef CAS PubMed.
- A. S. Schmidt and A. B. Thomsen, Bioresour. Technol., 1998, 64, 139–151 CrossRef CAS.
- E. Arvaniti, A. B. Bjerre and J. E. Schmidt, Biomass Bioenergy, 2012, 39, 94–105 CrossRef.
- S. Banerjee, R. Sen, R. A. Pandey, T. Chakrabarti, D. Satpute, B. S. Giri and S. Mudliar, Biomass Bioenergy, 2009, 33, 1680–1686 CrossRef CAS.
- C. A. Cardona, J. A. Quintero and I. C. Paz, Bioresour. Technol., 2010, 101, 4754–4766 CrossRef CAS PubMed.
- A. Kallioinen, M. Hakola, T. Riekkola, T. Repo, M. Leskela, N. von Weymarn and M. Siika-aho, Bioresour. Technol., 2013, 140, 414–420 CrossRef CAS PubMed.
- T. H. Kim and Y. Y. Lee, Bioresour. Technol., 2005, 96, 2007–2013 CrossRef CAS PubMed.
- S. P. S. Chundawat, L. Chang, C. Gunawan, V. Balan, C. McMahan and B. E. Dale, Ind. Crops Prod., 2012, 37, 486–492 CrossRef CAS.
- J. W. Kim, K. S. Kim, J. S. Lee, S. M. Park, H. Y. Cho, J. C. Park and J. S. Kim, Bioresour. Technol., 2011, 102, 8992–8999 CrossRef CAS PubMed.
- F. P. Bouxin, S. D. Jackson and M. C. Jarvis, Bioresour. Technol., 2014, 162, 236–242 CrossRef CAS.
- C. Zhao, W. Ding, F. Chen, C. Cheng and Q. Shao, Bioresour. Technol., 2014, 155, 34–40 CrossRef CAS PubMed.
- M. Gao, F. Xu, S. Li, S. Chen and D. Zhang, Biosystems Eng, 2010, 106, 470–475 CrossRef.
- N. Srinivasan and L. K. Ju, Bioresour. Technol., 2010, 101, 9785–9791 CrossRef CAS PubMed.
- K. H. Kim and J. Hong, Bioresour. Technol., 2001, 77, 139–144 CrossRef CAS PubMed.
- N. Narayanaswamy, A. Faik, D. J. Goetz and T. Gu, Bioresour. Technol., 2013, 102(13), 6995–7000 CrossRef PubMed.
- Y. Zeng, S. Zhao, S. Yang and S. Y. Ding, Curr. Opin. Biotechnol., 2014, 27, 38–45 CrossRef CAS PubMed.
- P. Alvira, E. T. Pejo, M. Ballesteros and M. J. Negro, Bioresour. Technol., 2010, 101, 4851–4861 CrossRef CAS PubMed.
- Y. Zeng, S. Zhao, S. Yang and S. Y. Ding, Curr. Opin. Biotechnol., 2014, 27, 38–45 CrossRef CAS PubMed.
- L. Zhu, J. P. O'Dwyer, V. S. Chang, C. B. Granda and M. T. Holtzapple, Bioresour. Technol., 2008, 99, 3817–3828 CrossRef CAS PubMed.
- J. A. Rollin, Z. Zhu, N. Sathitsuksanoh and Y. H. P. Zhang, Biotechnol. Bioeng., 2011, 108(1), 22–30 CrossRef CAS PubMed.
- S. Y. Ding, Y. S. Liu, Y. Zeng, M. E. Himmel, J. O. Baker and E. A. Bayer, Science, 2012, 338, 1055–1059 CrossRef CAS PubMed.
- T. D. H. Bugg, M. Ahmad, E. M. Hardiman and R. Rahmanpour, Nat. Prod. Rep., 2011, 28, 1883–1896 RSC.
- C. O. Boateng and K. T. Lee, Chem. Eng. J., 2013, 228, 162–171 CrossRef.
- A. Limayem and S. C. Ricke, Prog. Energy Combust. Sci., 2012, 38, 449–467 CrossRef CAS.
- C. Wan and Y. Li, Biotechnol. Adv., 2012, 30, 1447–1457 CrossRef CAS PubMed.
- D. Jalc, Agricultural Applications, ed. F. Kempken, Springer, Heidelberg, Berlin, 2002, ch. 2, p. 20 Search PubMed.
- A. D. Moreno, D. Ibarra, P. Alvira, E. Tomás-Pejó and M. Ballesteros, Crit. Rev. Biotechnol., 2015, 35(3), 342–354 CrossRef PubMed.
- D. W. S. Wong, Appl. Biochem. Biotechnol., 2009, 157, 174–209 CrossRef CAS.
- K. A. Jensen Jr, C. J. Houtman, Z. C. Ryan and K. E. Hammel, Appl. Environ. Microbiol., 2001, 67(6), 2705–2711 CrossRef CAS PubMed.
- S. J. A. van Kuijk, A. S. M. Sonnenberg, J. J. P. Baars, W. H. Hendriks and J. W. Cone, Biotechnol. Adv., 2015, 33, 191–202 CrossRef CAS PubMed.
- B. C. Saha, N. Qureshi, G. J. Kennedy and M. A. Cota, Int. Biodeterior. Biodegrad., 2016, 109, 29–35 CrossRef CAS.
- N. Jagmann and B. Philipp, J. Biotechnol., 2014, 184, 209–218 CrossRef CAS PubMed.
- L. R. Lynd, Annu. Rev. Energ. Environ., 1996, 21, 403–465 CrossRef.
- A. K. Chandel, B. C. M. Goncalves, J. L. Strap and S. S. da Silva, Crit. Rev. Biotechnol., 2015, 35(3), 281–293 CrossRef PubMed.
- E. Palmqvist and B. H. Hagerdal, Bioresour. Technol., 2000, 74, 17–24 CrossRef CAS.
- X. Yu, J. Zeng, Y. Zheng and S. Chen, Process Biochem., 2014, 49, 457–465 CrossRef CAS.
- D. S. Lee, S. G. Wi, S. J. Lee, Y. G. Lee, Y. S. Kim and H. J. Bae, Bioresour. Technol., 2014, 158, 239–247 CrossRef CAS PubMed.
- B. Wang, Y. H. Rezenom, K. C. Cho, J. L. Tran, D. G. Lee, D. H. Russell, J. J. Gill, R. Young and K. H. Chu, Bioresour. Technol., 2014, 161, 162–170 CrossRef CAS PubMed.
- F. B. Pereira, A. Romani, H. A. Ruiz, J. A. Teixeira and L. Domingues, Bioresour. Technol., 2014, 161, 192–199 CrossRef CAS PubMed.
- H. Ling, W. Teo, B. Chen, S. S. J. Leong and M. W. Chang, Curr. Opin. Biotechnol., 2014, 29, 99–106 CrossRef CAS PubMed.
- S. Elleuche, C. Schroder, K. Sahm and G. Antranikian, Curr. Opin. Biotechnol., 2014, 29, 116–123 CrossRef CAS PubMed.
- H. Ling, W. Teo, B. Chen, S. S. J. Leong and M. W. Chang, Curr. Opin. Biotechnol., 2014, 29, 99–106 CrossRef CAS PubMed.
- L. Viikari, J. Vehmaanpera and A. Koivula, Biomass Bioenergy, 2012, 46, 13–24 CrossRef CAS.
- A. Bhalla, N. Bansal, S. Kumar, K. M. Bischoff and R. K. Sani, Bioresour. Technol., 2013, 128, 751–759 CrossRef CAS PubMed.
- M. Basen, A. M. Rhaesa, I. Kataeva, C. J. Prybol, I. M. Scott, F. L. Poole and M. W. W. Adams, Bioresour. Technol., 2014, 152, 384–392 CrossRef CAS PubMed.
- A. Demirbas, Energy Sources, Part A, 2008, 30, 101–109 CrossRef CAS.
- M. J. Taherzadeh and K. Karimi, Int. J. Mol. Sci., 2008, 9, 1621–1651 CrossRef CAS PubMed.
- E Instruments, http://www.e-inst.com/biomass-to-biogas/, accessed February 2016.
- T. Bond and M. R. Templeton, Energy Sustainable Dev., 2011, 15, 347–354 CrossRef.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- D. L. Klass, Biomass for Renewable Energy, Fuels, and Chemicals, Academic Press, San Diego, 1998, pp. 272–275 Search PubMed.
- P. Basu, Biomass Gasification and Pyrolysis: Practical Design and Theory, Academic Press, United States, 2010 Search PubMed.
- Y. Lee, P. R. B. Eun, C. Ryu, Y. K. Park, J. H. Jung and S. Hun, Bioresour. Technol., 2013, 130, 345–350 CrossRef CAS PubMed.
- A. V. Bridgwater, D. Meier and D. Radlein, Org. Geochem., 1999, 30(12), 1479–1493 CrossRef CAS.
- A. Demirbas, Energy Sources, 2002, 24, 869–876 CrossRef CAS.
- J. D. Rocha, S. D. Brown, G. D. Love and C. E. Snape, J. Anal. Appl. Pyrolysis, 1997, 40–41, 91–103 CrossRef.
- Wikipedia, https://en.wikipedia.org/wiki/Fermentation, accessed February 2016.
- Scitable, http://www.nature.com/scitable/topicpage/yeast-fermentation-and-the-making-of-beer-14372813, accessed February 2016.
- J. L. Wertz and O. Bedue, Lignocellulosic Biorefineries, CRC Press, Spain, 2013, ch. 8, pp. 366–375 Search PubMed.
- B. H. Hagerdahl, K. Karhumaa, M. Jeppsson and M. F. G. Grauslund, Biofuels, ed. L. Olsson, Springer, Heidelberg, Berlin, 2007, pp. 147–177 Search PubMed.
- M. Ni, D. Y. C. Leung, M. K. H. Leung and K. Sumathy, Fuel Process. Technol., 2006, 87, 461–472 CrossRef CAS.
- V. M. Merino, M. J. Gil and A. Cornejo, Renewable Hydrogen Technologies: Production, Purification, Storage, Application and Safety, ed. L. M. Gandia, G. Arzamendi and P. M. Dieguez, Elsevier B. V., Poland, 2013, ch. 5, pp. 104–105 Search PubMed.
- P. Sivagurunathan, G. Kumar, P. Bakonyi, S. H. Kim, T. Kobayashi, K. Q. Xu, G. Lakner, G. Toth, N. Nemestothy and K. B. Bako, Int. J. Hydrogen Energy, 2016, 41, 3820–3836 CrossRef CAS.
- H. Zilouei and M. Taherdanak, Lignocellulose-Based Bioproducts, ed. K. Karimi, Springer International Publishing, Switzerland, 2015, ch. 7 Search PubMed.
- A. Ghimire, L. Frunzo, F. Pirozzi, E. Trably, R. Escudie, P. N. L. Lens and G. Esposito, Appl. Energy, 2015, 144, 73–95 CrossRef CAS.
- D. Karakashev and I. Angelidaki, Biofuels Alternative Feedstocks and Conversion Processes, ed. A. Pandey, C. Larroche, S. C. Ricke, C. G. Dussap and E. Gnansounou, Academic Press, USA, 2011, ch. 23, p. 527 Search PubMed.
- S. M. Kotay and D. Das, Int. J. Hydrogen Energy, 2008, 33, 258–263 CrossRef.
- V. M. Merino, M. J. Gil and A. Cornejo, Renewable Hydrogen Technologies: Production, Purification, Storage, Application and Safety, ed. L. M. Gandia, G. Arzamendi and P. M. Dieguez, Elsevier B. V., Poland, 2013, ch. 8, p. 180 Search PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfutzenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- P. K. Rout, A. D. Nannaware, O. Prakash, A. Kalra and R. Rajasekharan, Chem. Eng. Sci., 2016, 142, 318–346 CrossRef CAS.
- S. Peleteiro, S. Rivas, J. L. Alonso, V. Santos and J. C. Parajo, Bioresour. Technol., 2016, 202, 181–191 CrossRef CAS PubMed.
- J. P. Lange, E. van der Heide, J. van Buijtene and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- D. V. Hernandez, J. M. R. Caballero, J. S. Gonzalez, R. M. Tost, J. M. Robles, M. A. P. Cruz, A. J. Lopez, R. H. Huesca and P. M. Torres, J. Mol. Catal. A-Chem., 2014, 383–384, 106–113 CrossRef.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- M. I. Alam and B. Saha, Sustainable Catalytic Processes, ed. B. Saha, M. Fan and J. Wang, Elsevier, Amsterdam, 2015, ch. 4, p. 107 Search PubMed.
- S. Choi, C. W. Song, J. H. Shin and S. Y. Lee, Metab. Eng., 2015, 28, 223–239 CrossRef CAS PubMed.
- Y. R. Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–986 CrossRef PubMed.
- J. Jae, W. Zheng, R. F. Lobo and D. G. Vlachos, ChemSusChem, 2013, 6, 1158–1162 CrossRef CAS PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Green Chem., 2006, 8, 701–709 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520–540 RSC.
- J. Wisniak, M. Hershkowitz, R. Leibowitz and S. Stein, Ind. Eng. Chem. Prod. Res. Dev., 1974, 13(1), 75–79 CAS.
- A. Romero, E. Alonso, A. Sastre and A. N. Marquez, Microporous Mesoporous Mater., 2016, 224, 1–8 CrossRef CAS.
- G. van Ling and J. C. Vlugter, J. Appl. Chem., 1969, 19, 43–45 CrossRef CAS.
- I. T. Clark, Ind. Eng. Chem., 1958, 50(8), 1125–1126 CrossRef CAS.
- J. Sun and H. Liu, Green Chem., 2011, 13, 135–142 RSC.
- M. A. Andrews and S. A. Klaeren, J. Am. Chem. Soc., 1989, 111, 4133–4134 CrossRef.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- P. B. Smith, Biobased Monomers, Polymers, and Polymers, ed. P. B. Smith and R. A. Gross, Oxford University Press, Inc., United States of America, 2012, ch. 12, p. 186 Search PubMed.
- Y. Jiang, X. Wang, Q. Cao, L. Dong, J. Guan and X. Mu, Sustainable Production of Bulk Chemicals, ed. M. Xian, Springer, Dordrecht, Netherlands, 2016, ch. 2, p. 28 Search PubMed.
- R. P. John, K. M. Nampoothiri and A. Pandey, Appl. Microbiol. Biotechnol., 2007, 74, 524–534 CrossRef CAS PubMed.
- O. Kandler, Antonie van Leeuwenhoek, 1983, 49, 209–224 CrossRef CAS PubMed.
- H. Song and S. Y. Lee, Enzyme Microb. Technol., 2006, 39, 352–361 CrossRef CAS.
- J. G. Zeikus, M. K. Jain and P. Elankovan, Appl. Microbiol. Biotechnol., 1999, 51, 545–552 CrossRef CAS.
- D. P. Clark, FEMS Microbiol. Rev., 1989, 63, 223–234 CrossRef CAS.
- T. Yoshikawa, T. Yagi, S. Shinohara, T. Fukunaga, Y. Nakasaka, T. Tago and T. Masuda, Fuel Process. Technol., 2013, 108, 69–75 CrossRef CAS.
- M. Thevenot, M. F. Dignac and C. Rumpel, Soil Biol. Biochem., 2010, 42, 1200–1211 CrossRef CAS.
- S. Kang, X. Li, J. Fan and J. Chang, Renewable Sustainable Energy Rev., 2013, 27, 546–558 CrossRef CAS.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem.–Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- M. Kleinert and T. Barth, Chem. Eng. Technol., 2008, 31(5), 736–745 CrossRef CAS.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34(1), 29–41 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4(1), 35–46 CrossRef CAS.
- M. I. F. Mota, P. C. R. Pinto, J. M. Loureiro and A. E. Rodrigues, Sep. Purif. Rev., 2016, 45(3), 227–259 CrossRef CAS.
- L. Wang, G. Mu, C. Tan, L. Sun, W. Zhou, P. Yu, J. Yin and H. Fu, ChemSusChem, 2013, 6, 880–889 CrossRef CAS PubMed.
- G. Annadurai, R. S. Juang and D. J. Lee, J. Hazard. Mater., 2002, B92, 263–274 CrossRef.
- L. Brinchi, F. Contana, E. Fortunati and J. M. Kenny, Carbohydr. Polym., 2013, 94, 154–169 CrossRef CAS PubMed.
- J. Yang, K. Christiansen and S. Luchner, Renewable, Low-Cost Carbon Fiber for Lightweight Vehicles, U.S. Department of Energy, Detroit, 2013 Search PubMed.
- E. Lam, K. B. Male, J. H. Chong, A. C. W. Leung and J. H. T. Luong, Trends Biotechnol., 2012, 30(5), 283–290 CrossRef CAS PubMed.
- W. S. W. Ngah and M. A. K. M. Hanafiah, Bioresour. Technol., 2008, 99, 3935–3948 CrossRef PubMed.
- J. Febrianto, A. N. Kosasih, J. Sunarso, Y. H. Ju, N. Indraswati and S. Ismadji, J. Hazard. Mater., 2009, 162, 616–645 CrossRef CAS PubMed.
- B. Hu, K. Wang, L. Wu, S. H. Yu, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 1–16 CrossRef.
- C. Falco, J. M. Sieben, N. Brun, M. Sevilla, T. van der Mauelen, E. Morallon, D. C. Amoros and M. M. Titirici, ChemSusChem, 2013, 6, 374–382 CrossRef CAS PubMed.
- A. E. Ismanto, S. Wang, F. E. Soetaredjo and S. Ismadji, Bioresour. Technol., 2010, 101, 3534–3540 CrossRef CAS PubMed.
- F. Kurniawan, M. Wongso, A. Ayucitra, F. E. Soetaredjo, A. E. Angkawijaya, Y. H. Ju and S. Ismadji, J. Taiwan Inst. Chem. Eng., 2015, 197–201 CrossRef CAS.
- S. T. Senthilkumar, R. K. Selvan and J. S. Melo, AIP Conf. Proc., 2013, 1538, 124–127 CrossRef CAS.
- A. Funke and F. Ziegler, Biofuels, Bioprod. Biorefin., 2010, 4, 160–177 CrossRef CAS.
- J. M. Rosas, R. Berenguer, M. J. V. Romero, J. R. Mirasol and T. Cordero, Frontiers in Materials, 2014, 1, 1–17 CrossRef.
- S. A. Soenjaya, N. Handoyo, F. E. Soetaredjo, A. E. Angkawijaya, Y. H. Ju and S. Ismadji, International Journal of Industrial Chemistry, 2015, 6, 1–7 CrossRef.
- Y. Zhang, T. Nypelo, C. Salas, J. Arbodela, I. C. Hoeger and O. J. Rojas, J. Renewable Mater., 2013, 1(3), 195–211 CrossRef CAS.
- A. W. Carpenter, C. F. de Lannoy and M. R. Wiesnerm, Environ. Sci. Technol., 2015, 49(9), 5277–5287 CrossRef CAS PubMed.
- K. Spence, Y. Habibi and A. Dufresne, Bio- and Nano-Polymer Composites, ed. S. Kaila, B. S. Kaith and I. Kaur, Springer, Berlin, 2011 Search PubMed.
- I. Siro and D. Plackett, Cellulose, 2010, 17, 459–494 CrossRef CAS.
- M. Henriksson, G. Henriksson, L. A. Berglund and T. Lindstrom, Eur. Polym. J., 2007, 43, 3434–3441 CrossRef CAS.
- N. Lavoine, I. Desloges, A. Dufresne and J. Bras, Carbohydr. Polym., 2012, 90, 735–764 CrossRef CAS PubMed.
- H. P. Zhao, X. Q. Feng and H. Gao, Appl. Phys. Lett., 2007, 90, 73112–73114 CrossRef.
- P. B. Filson and B. E. D. Andoh, Bioresour. Technol., 2009, 100, 2259–2264 CrossRef CAS PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS.
- J. Araki, M. Wada, S. Kuga and T. Okano, Colloids Surf., A, 1998, 142, 75–82 CrossRef CAS.
- P. Lu and Y. L. Hsieh, Carbohydr. Polym., 2010, 82, 329–336 CrossRef CAS.
- Y. Peng, D. J. Gardner and Y. Han, Cellulose, 2012, 19, 91–102 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |