
Novel A–(π–D–π–A)1–3 branched fluorophores displaying high two-photon absorption†
Abstract
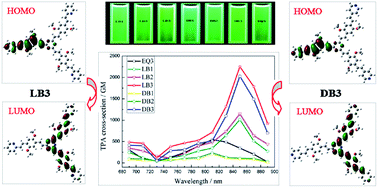
A series of novel acceptor–π–donor–π–acceptor branched fluorophores with a 1,3,5-triazine core (LB1, LB2, LB3, DB1, DB2, DB3, and EQ3) were synthesized and characterized by infrared, hydrogen-1 nuclear magnetic resonance, carbon-13 nuclear magnetic resonance, mass spectrometry, and elemental analysis. Their photophysical properties in different solvents were systematically investigated including linear absorption, single-photon excited fluorescence, two-photon absorption, and frequency up-converted fluorescence. When the number of branches increases, the spectral positions of the linear absorption and the single-photon excited fluorescence show red shifts, while the fluorescence quantum yields decrease. When the polarity of solvents increases, the spectral positions of the single-photon excited fluorescence and the Stokes shifts also show red shifts, while the linear absorption wavelengths slightly change and the fluorescence quantum yields decrease. Under the excitation of a femtosecond laser (690–890 nm, 80 MHz, 140 fs), all these compounds emit intense green frequency up-converted fluorescence, and the two-photon absorption cross-sections in THF are 236, 1149, 2250, 207, 1015, 2028, and 546 GM for LB1, LB2, LB3, DB1, DB2, DB3, and EQ3, respectively. The quantum chemical calculations based on density functional theory provide supporting evidence that significant enhancement of the two-photon absorption cross-section can be achieved by sufficient electronic coupling between the strong charge transfer acceptor–π–donor–π–acceptor quadrupolar branches through the 1,3,5-triazine core.
 Please wait while we load your content...
Please wait while we load your content...

