
Carbon nanotube-mediated delivery of budesonide to macrophages†
Stéphanie Foillarda,
Julie Russierb,
Cécile Seifertb,
Hélène Dumortier*b and
Eric Doris*a
aCEA, iBiTecS, Service de Chimie Bioorganique et de Marquage, 91191 Gif-sur-Yvette, France. E-mail: eric.doris@cea.fr
bCNRS, Immunopathologie et Chimie Thérapeutique/Laboratory of Excellence Medalis, Institut de Biologie Moléculaire et Cellulaire, 67084 Strasbourg, France. E-mail: h.dumortier@ibmc-cnrs.unistra.fr
First published on 20th May 2016
Abstract
Carbon nanotubes were functionalized with budesonide, a potent anti-inflammatory drug, and investigated for interactions with macrophages. Pristine carbon nanotubes were first shortened to make them compatible with cellular dimensions before budesonide was non-covalently appended to their surface via a specifically designed amphiphilic molecule. The resulting nanohybrids were evaluated in regards to their internalisation into macrophages and ability to inhibit pro-inflammatory cytokine secretion. In vitro experiments showed that the budesonide delivery system is readily captured by phagocytic cells and has a deep anti-inflammatory effect as it strongly inhibits IL-6 production.
Introduction
Budesonide (BUD) is a synthetic glucocorticoid with a potent local anti-inflammatory effect which however, exhibits weak systemic activity due to extensive first-pass hepatic metabolism. BUD is currently used for the treatment of chronic inflammatory diseases such as asthma (through inhalation),1 Crohn's disease or ulcerative colitis2 (through rectal, oral or intravenous routes). After cellular internalization, BUD binds the cytoplasmic glucocorticoid receptor (GR), which triggers the dissociation from chaperone proteins and translocation of the BUD–GR complex to the nucleus. There, BUD–GR not only activates the transcription of genes that code for anti-inflammatory molecules but also inhibits major transcription factors such as NFκB and AP-1. The latter classically control the expression of most pro-inflammatory-cytokines.3 Although efficient, BUD is a highly hydrophobic drug, which is mostly insoluble in aqueous medium. As a result, its bioavailability is limited and high doses of the drug are required, which in turn lead to side effects and systemic toxicity that outweigh benefits to long-term treated patients.4 These intrinsic problems associated to BUD and other corticoids may be overcome by using nanometric formulations.5 Indeed, nano-sized carriers have the ability to improve the pharmacological and therapeutic profile of a given drug as the carrier system is able to protect the drug from the surrounding medium, enhance its aqueous solubility, and carry the drug to its therapeutic target.6 As the latter property could lead to an increased specificity, one can expect lower administered doses and therefore fewer side effects.The ability of some nanocarriers to passively accumulate in inflamed tissue and be taken up by macrophages has already been successfully exploited for the formulation of anti-inflammatory drugs.7 Indeed, local inflammation leads to micro environment changes and more permeable blood vessels.8 Accordingly, nanocarriers can leave the vascular bed and reach the interstitial space as demonstrated, for example, with PLGA nanoparticles or liposomes.9 As part of our long-standing interest in carbon nanotubes (CNT) for biomedical10 and other applications,11 we sought to exploit the carbonaceous platform for the delivery of anti-inflammatory BUD to macrophages. Indeed, these cells are central in the development of pro-inflammatory processes and induction of immune responses. Macrophages are phagocytic cells located in most tissues and are part of the first cellular line of defense of the body.12 Therefore they are likely to rapidly encounter and capture foreign particles upon exposure, which makes CNT promising candidates for the acute delivery of anti-inflammatory drugs.13 We report here our strategy for the construction of a BUD–CNT nanohybrid and its evaluation in the delivery of the anti-inflammatory drug to macrophages.
Results and discussion
Carbon nanotubes are tubular-shaped structures made from the rolling-up of graphene sheets into cylinders. CNT have already been used for the shuttling of various active compounds such as, proteins, siRNA, and small drug molecules both in vitro and in vivo.14 The carbonaceous platform offers some advantages over other carriers in that it possesses high specific surface area, thus allowing the cargoeing of large quantities of active compounds. Our BUD–CNT delivery system was designed taking four important aspects into consideration: (i) aqueous dispersibility of the CNT-conjugate, (ii) reduced side cytotoxicity of the functionalized CNT, (iii) protection of the drug from the surroundings, (iv) ability of the carrier system to release the drug at some point to allow pharmacological effects.Our approach is based on the non-covalent functionalization of CNT by self-assembly of the amphiphilic compound 1 (Scheme 1). The latter is made of a lipophilic chain connected to a polyethyleneglycol (MW 2000, PEG2000) polar head. The amphiphilic unit also incorporates an ester side chain linked to a budesonide molecule. The choice of an ester linkage was governed by the latent release of budesonide under mild conditions. While the hydrophobic portion of the amphiphile is expected to adsorb on the surface of the CNT by van der Waals interactions, its polar head will orientate towards the aqueous solution. The polyether unit (PEG2000) is anticipated to confer the requested aqueous solubility to the CNT and shield the drug.
 | ||
| Scheme 1 Synthetic route to budesonide-based amphiphile 1. | ||
Synthesis of the budesonide-amphiphile (BUD-amphiphile)
The synthesis of 1 started from masked glutamic acid 2 which was used as central connecting point for the three different components of the amphiphile, i.e. lipophilic chain, hydrophobic chain, and budesonide (Scheme 1). The carboxylic group of 2 was first reacted with octadecylamine to afford lipophilic compound 3 whose Fmoc group was removed using piperidine. The resulting amino group was subsequently engaged in an amidation reaction of MeO-PEG2000-COOH to deliver compound 4. The side tert-butyl group of 4 was then deprotected and the ensuing carboxylic acid finally esterified with the primary alcohol group of budesonide using DCC as activating agent. Compound 1 was obtained in five linear steps in ca. 5% overall yields. The above scheme permitted to design an amphiphile in which the budesonide molecule is embedded in-between the lipophilic and hydrophobic regions and thus protected from the surroundings. Amphiphile 1 is expected to play a dual role, namely solubilizing group for carbon nanotubes and anchoring unit for budesonide. With amphiphile 1 in hands, we next investigated its adsorption properties on the surface of carbon nanotubes.Functionalization of carbon nanotubes
Macrophages can engulf nanomaterials of various sizes and shapes including carbon nanotubes, but exposure to long CNT could result in frustrated phagocytosis and induce the release of pro-inflammatory cytokines.15 As the uptake of CNT into macrophages is dependent on their length and degree of bundling (aggregation), we conjectured that small CNT of a few hundreds of nanometers in length would be more compatible with cellular dimensions. Hence, commercially available multiwalled CNT (1–3 μm long) were dispersed in toluene and shortened by extensive ultrasonication. Transmission electron microscopy (TEM) analyses indicated that ca. 80% of the nanotubes were shortened to lengths around 200 nm. The shortened CNT were then dispersed in H2O by sonication at 0 °C in the presence of the amphiphile 1 synthesized above. Upon interaction with 1, the CNT became spontaneously suspended in water (Fig. 1a).‡ The CNT–BUD nanohybrid was subsequently collected by centrifugation, washed several times with water to remove amphiphile in excess, and characterized using standard analytical methods. TEM analysis showed short CNT with no morphological alteration and the CNT–BUD were mostly found as individual species (Fig. 1b). X-Ray photoelectron spectroscopy (XPS) permitted to quantify the budesonide content as we observed an increase (relative to the oxygen content of the shortened CNT) of the O 1s peak at 531 eV originating from the amphiphile's oxygen atoms (Fig. 1c). From the oxygen-to-carbon ratio, we calculated that the nanohybrid contained about 18 wt% of the amphiphile, which corresponds to ca. 3 wt% of budesonide.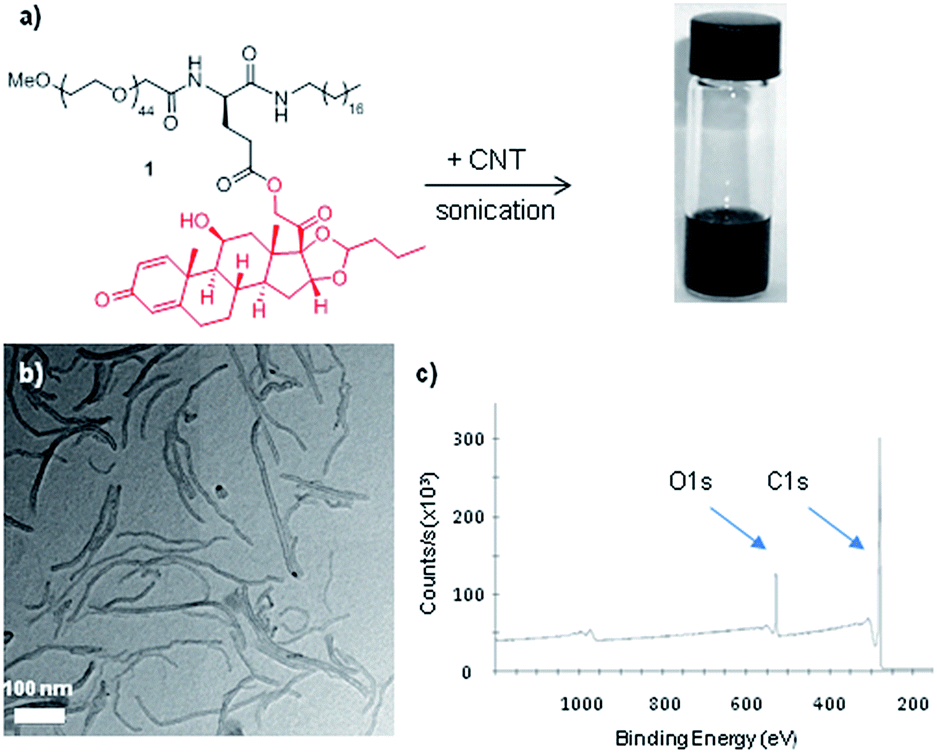 | ||
| Fig. 1 (a) Functionalization of carbon nanotubes with amphiphile 1 resulting in the aqueous dispersion of CNTs. (b) TEM image of the budesonide-functionalized carbon nanotubes. (c) XPS of the budesonide-functionalized carbon nanotubes. | ||
CNT–BUD uptake by macrophages
The budesonide nanohybrid was studied in interaction with macrophages as regards its cellular uptake. Thus, CNT–BUD was incubated for 24 h with either RAW 264.7 macrophages (mouse cell line) or primary peritoneal murine macrophages. The internalization process was first monitored by optical microscopy. As controls, cells were either left untreated or incubated with BUD-free CNT (CNT-CTRL), i.e. CNT simply dispersed with a pegylated amphiphile (5) incorporating no BUD (see ESI† for details). As shown in Fig. 2, both types of nanotubes, i.e. CNT-CTRL and CNT–BUD, seems to be internalized in a “dose-dependent” manner by the two types of macrophages as we visually observed more intracellular carbon nanotubes in the “high concentration” (34 μg mL−1 of CNT) experiment compared to the “medium concentration” (3.4 μg mL−1 of CNT) experiment. Although the above experiment did not unambiguously tell whether CNT were located inside the cells, it was a first indication that they were cell-associated and not floating in the medium. This was further confirmed by electron microscopy (see below). | ||
| Fig. 2 Visualization of CNT uptake by (A) the RAW 264.7 macrophage cell line and (B) primary mouse peritoneal macrophages, using optical microscopy (×200). Cells were either left untreated (a) or incubated for 24 h with CNT–BUD at high concentration (b) (2 μM BUD, 34 μg mL−1 CNT) or medium concentration (c) (0.2 μM BUD, 3.4 μg mL−1 CNT), or with CNT-CTRL at high (d) or medium (e) concentration. | ||
Further investigation by TEM allowed us to visualize more into details the exact intracellular localization of the CNT-based nanohybrids. With the final aim of performing functional and physiologically relevant analyses (see next paragraph & Fig. 4), we incubated lipopolysaccharides (LPS)-activated primary macrophages with CNT-CTRL or CNT–BUD (50 μg mL−1) for 24 h. TEM micrographs (Fig. 3) indicated that large CNT bundles were mainly located inside phagocytic vesicles with little to no effect on the morphology of the cells. Interestingly, magnified TEM images (Fig. 3e and f) showed some individualized CNT in the cytoplasm and more importantly in the cell nucleus. This suggests that the nanohybrid is able, to some extent, to escape phagosomes. It could then reach the cytoplasmic glucocorticoid receptor and translocate to the nucleus. The latter observation is of key importance as to our final objective given that the BUD-induced transcriptional down-regulation of genes involved in the production of pro-inflammatory cytokines takes place in the nucleus.3 In addition, budesonide itself is likely to escape from the phagosomes as the ester linkage connecting the anti-inflammatory drug to the nanotube platform is labile under mildly acidic conditions such as the ones found in the phagosomal compartment. Thus, BUD can also be released from CNT–BUD after internalisation and further migrate to the nucleus.
 | ||
| Fig. 3 Visualization of intracellular localization of CNT upon uptake by primary mouse peritoneal macrophages using TEM. Cells were either left untreated (a), stimulated with LPS (t = 24 h) (b), incubated with LPS and CNT-CTRL (t = 24 h) (c), or incubated with LPS and CNT–BUD (t = 24 h) (d). Areas surrounded by dotted or continuous lines in (d) are enlarged in (e) and (f) respectively. N, nucleus. Representative images of 5–15 cells analysed per condition. | ||
Our data demonstrate that the CNT–BUD nanohybrid is easily and rapidly captured by phagocytic cells and is likely to reach its receptor location. This result prompted us to evaluate its anti-inflammatory potential by analysing its capacity in inhibiting the production of interleukin 6 (IL-6), a pro-inflammatory cytokine.
Assessment of the anti-inflammatory potential of CNT–BUD
We first analyzed the potential intrinsic pro-inflammatory properties of the carbonaceous carrier as CNT have been described, in some studies, to spontaneously trigger inflammatory responses.16 As shown in Fig. 4, neither CNT-CTRL nor CNT–BUD (50 μg mL−1) induce the release of IL-6 by primary murine macrophages, which indicates they do not induce inflammation themselves. On the contrary, LPS triggers macrophage activation, which translates into strong IL-6 secretion. We thus assessed the ability of the CNT-bonded drug to inhibit such LPS-induced IL-6 production by macrophages. For this purpose, murine macrophages were incubated with LPS in the presence of CNT–BUD. The anti-inflammatory properties of CNT–BUD were compared to those of BUD alone and BUD formulated into micelles made from the spontaneous self-assembly of amphiphile 1 in the absence of CNT (Mic–BUD). Indeed, micelles can behave as nanometric cargos and have been extensively used in the literature for drug delivery applications.17 Empty micelles incorporating no BUD (Mic-CTRL) were also assembled from a simple pegylated amphiphile 5. Results are depicted in Fig. 4. Medium and low concentrations of BUD, CNT-CTRL (without budesonide), CNT–BUD (with budesonide), Mic-CTRL (without budesonide) and Mic–BUD (with budesonide) were incubated with LPS-activated macrophages for 24 h. As expected, little to no inhibition of IL-6 secretion was observed in the presence of CNT-CTRL or Mic-CTRL. On the contrary, IL-6 secretion was highly inhibited (>95% inhibition) when BUD was added to the cell culture in the 0.02–0.2 μM range. Interestingly, the CNT–BUD nanohybrid was also able to drastically inhibit the release of IL-6 (>95%) at the two investigated concentrations, including the lowest one (0.34 μg mL−1 of CNT–BUD corresponding to 0.02 μM of BUD). Mic–BUD also had a significant but weaker influence on IL-6 production as >95% inhibition was observed at 0.2 μM but only ca. 70% inhibition was measured at 0.02 μM. Taken together, these results demonstrate that the nanotube-based formulation of BUD does not alter the potency of the drug as the hybrid exhibit high anti-inflammatory effect at the concentrations studied. Lower concentrations tested did not allow us to discriminate between BUD and CNT–BUD efficiencies (data not shown). The performances of CNT–BUD can be rationalized by its rapid phagocytosis and the improvement of aqueous solubility of the drug when coupled to CNT. The latter phenomenon is also likely impacting its bioavailability which, in turn, should lead to less adverse effects upon in vivo administration as lower doses will be required.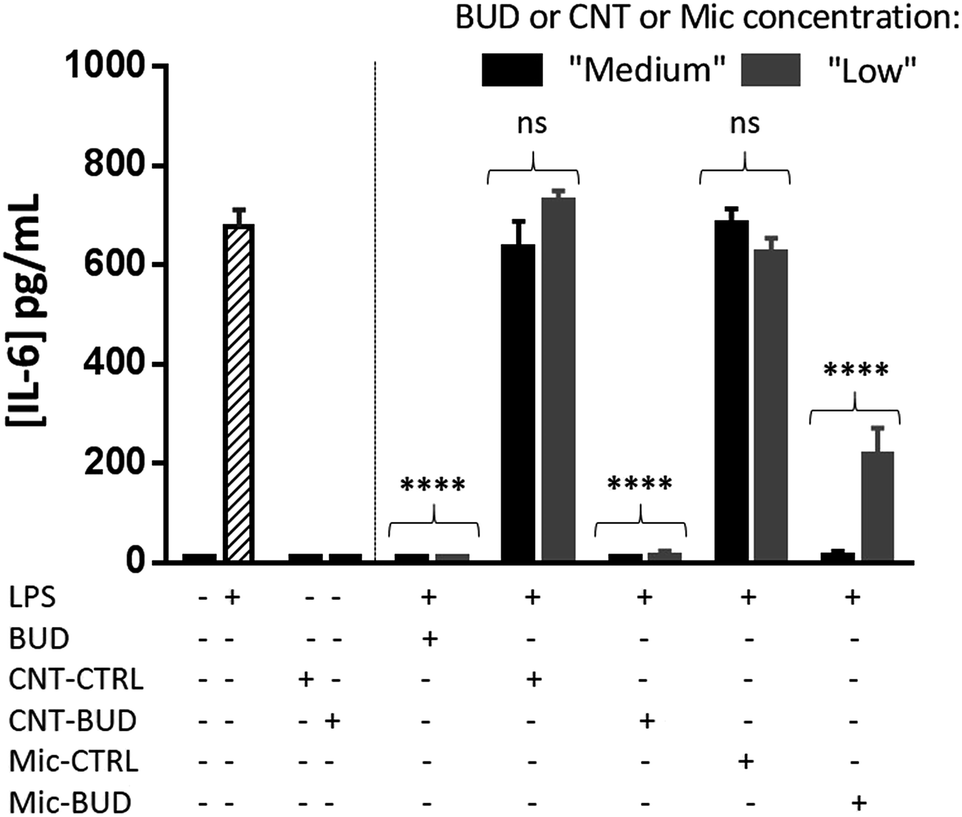 | ||
| Fig. 4 Secretion of the pro-inflammatory cytokine IL-6 by primary mouse peritoneal macrophages (ELISA test, t = 24 h). Left side: macrophages were either left untreated or stimulated with LPS (20 ng mL−1) or cultured with CNT-CTRL or CNT–BUD (50 μg mL−1). Right side: macrophages were stimulated with LPS (20 ng mL−1) together with BUD, CNT-CTRL, CNT–BUD, Mic-CTRL or Mic–BUD, at two different concentrations. Black bars: medium concentration (0.2 μM BUD or 3.4 μg mL−1 CNT or 1.1 μg mL−1 Mic); grey bars: low concentration (0.02 μM BUD or 0.34 μg mL−1 CNT or 0.11 μg mL−1 Mic). Results are expressed as mean concentration +/− SD from one representative experiment out of 2–4. ****p < 0.0001 (one-way Anova, compared to the LPS activation condition, hatched bar). ns, not significant. | ||
Experimental
Mice were maintained in our animal facility (approved by French Veterinary Services, #F67-482-2); experiments were carried out in conformity with the 2010/63/UE European animal bioethics legislation.Synthesis of the budesonide-amphiphile (BUD-amphiphile 1)
Deprotection of the Fmoc group. The above prepared Fmoc-Glu(OtBu)-NH-C18H35 3 (0.176 g, 0.26 mmol) was stirred in piperidine/DMF 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 (10 mL) at room temperature for 4 h. The solvent was removed under vacuum and the crude product was purified by column chromatography on silica gel (eluent: CH2Cl2/MeOH, 98/2) to yield the desired amino-deprotected NH2-Glu(OtBu)-NH-C18H35 3′ as a colorless oil (63% yield). 1H NMR (CDCl3) δ 0.85–0.91 (t, 3H, J = 6.8 Hz), 1.20–1.35 (m, 30H), 1.44 (s, 9H), 1.46–1.55 (m, 2H), 1.80–2.16 (m, 2H), 2.30–2.45 (m, 2H), 3.19–3.30 (m, 2H), 3.47–3.65 (m, 1H), 7.27–7.33 (m, 1H). HRMS calc. for C27H55N2O3 (M + H)+ 455.4213, found: 455.4203.
:8 (10 mL) at room temperature for 4 h. The solvent was removed under vacuum and the crude product was purified by column chromatography on silica gel (eluent: CH2Cl2/MeOH, 98/2) to yield the desired amino-deprotected NH2-Glu(OtBu)-NH-C18H35 3′ as a colorless oil (63% yield). 1H NMR (CDCl3) δ 0.85–0.91 (t, 3H, J = 6.8 Hz), 1.20–1.35 (m, 30H), 1.44 (s, 9H), 1.46–1.55 (m, 2H), 1.80–2.16 (m, 2H), 2.30–2.45 (m, 2H), 3.19–3.30 (m, 2H), 3.47–3.65 (m, 1H), 7.27–7.33 (m, 1H). HRMS calc. for C27H55N2O3 (M + H)+ 455.4213, found: 455.4203.
Coupling of the PEG unit. To a solution of NH2-Glu(OtBu)-NH-C18H35 (0.072 g, 0.16 mmol) (3′, prepared above) in DMF (10 mL) was added MeO-PEG2000-COOH (0.350 g, 0.17 mmol), DIPEA (65 μL, 0.37 mmol) and PyBOP (0.123 g, 0.24 mmol). The reaction mixture was stirred at room temperature for 16 h. The solvent was removed under vacuum and the crude product was purified by column chromatography on silica gel (eluent: CH2Cl2/MeOH, 98/2) to yield the desired MeO-PEG2000-CO-Glu(OtBu)-NH-C18H35 4 as a waxy solid (38% yield). 1H NMR (CDCl3) δ 0.85–0.90 (t, 3H, J = 6.8 Hz), 1.22–1.30 (m, 30H), 1.44 (s, 9H), 1.46–1.53 (m, 2H), 1.85–2.16 (m, 2H), 2.23–2.45 (m, 2H), 3.15–3.30 (m, 2H), 3.38 (s, 3H), 3.44–3.85 (m, HPEG), 3.95–4.08 (m, 2H), 4.38–4.46 (m, 1H), 6.40 (s, 1H), 7.41–7.48 (m, 1H). ESI-MS: m/z 811 (M + 2H + Na)3+.
Deprotection of the tert-butyl group. The above prepared MeO-PEG2000-CO-Glu(OtBu)-NH-C18H35 4 (0.146 g, 0.06 mmol) was stirred at room temperature for 3 h in TFA/CH2Cl2 50
:50 (5 mL). The solvent was removed under vacuum to yield the MeO-PEG2000-CO-Glu-NH-C18H35 4′ as a waxy solid (quantitative yield). 1H NMR (CDCl3) δ 0.85–0.90 (t, 3H, J = 6.8 Hz), 1.17–1.25 (m, 30H), 1.43–1.52 (m, 2H), 2.12–2.21 (m, 2H), 2.29–2.57 (m, 2H), 3.15–3.26 (m, 2H), 3.36 (s, 3H), 3.43–3.85 (m, HPEG), 4.02 (s, 2H), 4.16–4.20 (m, 1H), 6.40 (s, 1H), 7.41–7.48 (s, 1H). ESI-MS: m/z 1192 (M + H + Na)2+.
Coupling of budesonide. To a solution of MeO-PEG2000-CO-Glu-NH-C18H35 (100 mg, 0.042 mmol) (4′, prepared above) in anhydrous CH2Cl2 (5 mL) were added budesonide (18 mg, 0.042 mmol) and DMAP (2 mg, 0.017 mmol). The solution was cooled at 0 °C before the addition of DCC (16 mg, 0.063 mmol). The reaction mixture was stirred at room temperature for 24 h. The solvent was removed under vacuum and the crude product was purified by column chromatography on silica gel (eluent: CH2Cl2/MeOH, 99/1) to yield the desired MeO-PEG2000-CO-Glu(BUD)-NH-C18H35 1 as a waxy solid (47% yield). 1H NMR (CDCl3) δ 0.85–0.90 (t, 3H, J = 6.8 Hz), 0.90–1.10 (m, 6H), 1.22–1.30 (m, 30H), 1.45 (s, 3H), 1.07–1.92 (m, 15H), 1.98–2.07 (m, 2H), 2.10–2.20 (m, 2H), 2.50–2.62 (m, 2H), 3.16–3.32 (m, 3H), 3.38 (s, 3H), 3.53–3.85 (m, HPEG), 3.95 (s, 2H), 3.98–4.05 (m, 1H), 4.43–4.54 (m, 1H), 4.68–4.76 (m, 1H), 5.06–5.18 (m, 2H), 6.01–6.05 (m, 1H), 6.25–6.31 (m, 1H), 6.60–6.85 (m, 1H), 7.23–7.32 (s, 1H), 7.40–7.50 (s, 1H). ESI-MS: m/z 928 (M + 3H)3+.
Synthesis of MeO-PEG2000-CO-NH-C18H35 amphiphile (5)
Amphiphile 5 (structure depicted in the ESI†) is an analog of amphiphile 4 (see above) but without the budesonide side chain. To a solution of octadecylamine (33 mg, 0.12 mmol) in DMF (20 mL) was added MeO-PEG2000-COOH (200 mg, 0.10 mmol). DIPEA (38 μL, 0.22 mmol) and PyBOP (63 mg, 0.12 mmol) were then added. The reaction mixture was stirred at room temperature for 4 h. The solvent was removed under vacuum and the crude product was purified by column chromatography on silica gel (eluent: CH2Cl2/MeOH, 99/1) to yield the desired MeO-PEG2000-CO-NH-C18H35 amphiphile 5 as a colorless oil (57% yield). 1H NMR (CDCl3) δ 0.86–0.90 (t, 3H, J = 7 Hz), 1.22–1.35 (m, 30H), 1.48–1.54 (m, 2H), 3.25–3.31 (m, 2H), 3.38 (s, 3H), 3.45–3.85 (m, HPEG), 3.99 (s, 2H). ESI-MS: m/z 565 (M + 3H + Na)4+.Conclusions
Water dispersible BUD–CNT were successfully prepared by the assembly of BUD-containing amphiphiles on shortened nanotubes. The resulting nanohybrids were shown to be intrinsically non pro-inflammatory and readily taken-up by macrophages. In vitro experiments showed that the newly-designed BUD delivery system has a deep anti-inflammatory effect as it strongly inhibits IL-6 production by macrophages. CNT–BUD displays some advantages over BUD itself as it improves the solubility of hydrophobic BUD. This could allow the administration of lower doses of BUD, higher local bioavailability, and weaker in vivo systemic adverse effects. In addition, CNT–BUD is readily captured by macrophages which are the cells to target when one uses anti-inflammatory drugs. Upon in vivo administration, the nanohybrids are thus likely to be taken-up with priority by macrophages and other phagocytic cells, and more active formulations could even be conceived by using specific ligands to trigger, in addition to passive targeting, active delivery to the target.18Acknowledgements
This project was partly funded by MEDITRANS, an Integrated Project funded by the European Commission under the “nanotechnologies and nano-sciences, knowledge-based multifunctional materials and new production processes and devices” (NMP), thematic priority of the Sixth Framework Program. The “Service de Chimie Bioorganique et de Marquage” belongs to the Laboratory of Excellence in Research on Medication and Innovative Therapeutics (ANR-10-LABX-0033-LERMIT). TEM images of macrophages were recorded at the Microscopy Facility Platform of Esplanade Campus (Strasbourg, France).Notes and references
- K. M. Hvizdos and B. Jarvis, Drugs, 2000, 60, 1141 CrossRef CAS PubMed.
- (a) A. C. Ford, C. N. Bernstein, K. J. Khan, M. T. Abreu, J. K. Marshall, N. J. Talley and P. Moayyedi, Am. J. Gastroenterol., 2011, 106, 590 CrossRef CAS PubMed; (b) C. De Cassan, G. Fiorino and S. Danese, Dig. Dis., 2012, 30, 368 CrossRef PubMed.
- (a) M. Linden and R. Brattsand, Pulm. Pharmacol., 1994, 7, 43 CrossRef CAS PubMed; (b) P. J. Barnes, Immunol. Allergy Clin. North Am., 2005, 25, 451 CrossRef PubMed; (c) Y. Chinenov, R. Gupte and I. Rogatsky, Mol. Cell. Endocrinol., 2013, 380, 55 CrossRef CAS PubMed.
- H. Schäcke, W. D. Döcke and K. Asadullah, Pharmacol. Ther., 2002, 96, 23 CrossRef.
- A. Beloqui, R. Coco, M. Alhouayek, M. A. Solinis, A. Rodriguez-Gascon, G. G. Muccioli and V. Préat, Int. J. Pharm., 2013, 454, 775 CrossRef CAS PubMed.
- J. Ogier, T. Arnauld and E. Doris, Future Med. Chem., 2009, 1, 693 CrossRef CAS PubMed.
- (a) W. Ubrich and A. Lamprecht, J. R. Soc., Interface, 2010, 7, S55 CrossRef PubMed; (b) A. Lamprecht, N. Ubrich, H. Yamamoto, U. Schäfer, H. Takeuchi, P. Maincent, Y. Kawashima and C. M. Lehr, J. Pharmacol. Exp. Ther., 2001, 299, 775 CAS; (c) A. N. Ilinskaya and M. A. Dobrovolskaia, Br. J. Pharmacol., 2014, 171, 3988 CrossRef CAS PubMed; (d) G. Lamanna, J. Russier, H. Dumortier and A. Bianco, Biomaterials, 2012, 33, 5610 CrossRef CAS PubMed.
- J. Shaji and M. Lal, Asian J. Pharm. Clin. Res., 2013, 6(suppl. 3), 3 CAS.
- F. Leonard, H. Ali, E. M. Collnot, B. J. Crielaard, T. Lammers, G. Storm and C. M. Lehr, ALTEX, 2012, 29, 275 CrossRef PubMed.
- (a) S. Foillard, G. Zuber and E. Doris, Nanoscale, 2011, 3, 1461 RSC; (b) A. K. Varkouhi, S. Foillard, T. Lammers, R. M. Schiffelers, E. Doris, W. E. Hennink and G. Storm, Int. J. Pharm., 2011, 416, 419 CrossRef CAS PubMed; (c) C. Canapè, S. Foillard, R. Bonafè, A. Maiocchi and E. Doris, RSC Adv., 2015, 5, 68446 RSC.
- E. Gravel, I. N. N. Namboothiri and E. Doris, Synlett, 2016, 27, 1179 CrossRef CAS.
- L. C. Davies and P. R. Taylor, Immunology, 2015, 144, 541 CrossRef CAS PubMed.
- (a) G. D. Vuković, S. Z. Tomić, A. D. Marinković, V. Radmilović, P. S. Uskoković and M. Čolić, Carbon, 2010, 48, 3066 CrossRef; (b) X. Luo, C. Matranga, S. Tan, N. Alba and X. T. Cui, Biomaterials, 2011, 32, 6316 CrossRef CAS PubMed; (c) N. Lodhi, N. K. Mehra and N. K. Jain, J. Drug Targeting, 2013, 21, 67 CrossRef CAS PubMed.
- (a) A. Bianco and M. Prato, Adv. Mater., 2003, 15, 1765 CrossRef CAS; (b) A. Bianco, K. Kostarelos, C. D. Partidos and M. Prato, Chem. Commun., 2005, 5, 571 RSC; (c) M. Prato, K. Kostarelos and A. Bianco, Acc. Chem. Res., 2008, 41, 60 CrossRef CAS PubMed; (d) A. K. Bhaskar, V. N. Deshmukh and L. Prajapati, Int. J. Pharm. Technol., 2013, 5, 2695 CAS.
- M. S. Boyles, L. Young, D. M. Brown, L. MacCalman, H. Cowie, A. Moisala, F. Smail, P. J. Smith, L. Proudfoot, A. H. Windle and V. Stone, Toxicol. In Vitro, 2015, 29, 1513 CrossRef CAS PubMed.
- (a) H. Dumortier, Adv. Drug Delivery Rev., 2013, 65, 2120 CrossRef CAS PubMed; (b) K. Bhattacharya, Adv. Drug Delivery Rev., 2013, 65, 2087 CrossRef CAS PubMed.
- (a) M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343 CrossRef CAS PubMed; (b) U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomedicine, 2010, 6, 714 CAS; (c) T. Musacchio and V. P. Torchilin, Drugs Pharm. Sci., 2010, 194, 260 CAS; (d) S.-W. Kim, Y.-Z. Shi, J.-Y. Kim, K.-N. Park and J.-X. Cheng, Expert Opin. Drug Delivery, 2010, 7, 49 CrossRef CAS PubMed; (e) J. Ogier, T. Arnauld, G. Carrot, A. Lhumeau, J.-M. Delbos, C. Boursier, O. Loreau, F. Lefoulon and E. Doris, Org. Biomol. Chem., 2010, 8, 3902 RSC; (f) N. Mackiewicz, E. Gravel, A. Garofalakis, J. Ogier, J. John, D. M. Dupont, K. Gombert, B. Tavitian, E. Doris and F. Ducongé, Small, 2011, 19, 2786 CrossRef PubMed; (g) E. Gravel, J. Ogier, T. Arnauld, N. Mackiewicz, F. Ducongé and E. Doris, Chem.–Eur. J., 2012, 18, 400 CrossRef CAS PubMed; (h) E. Gravel, B. Thézé, I. Jacques, A. Parambath, K. Gombert, F. Ducongé and E. Doris, Nanoscale, 2013, 5, 1955 RSC; (i) A. Parambath, E. Gravel, I. Theodorou, K. Gombert, B. Thézé, F. Ducongé and E. Doris, Adv. Funct. Mater., 2014, 24, 5246 CrossRef; (j) Z. Ahmad, A. Shah, M. Siddiq and H. B. Kraatz, RSC Adv., 2014, 4, 17028 RSC; (k) I. Theodorou, A. Parambath, B. Lelandais, D. Clarisse, A. Doerflinger, E. Gravel, F. Ducongé and E. Doris, Chem. Commun., 2015, 51, 14937 RSC; (l) E. A. Rainbolt, K. E. Washington, M. C. Biewer and M. C. Stefan, Polym. Chem., 2015, 6, 2369 RSC.
- C. Kelly, C. Jefferies and S. A. Cryan, J. Drug Delivery, 2011, 727241 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra09809f |
| ‡ Direct adsorption of the drug on the nanotube was attempted by mixing authentic budesonide with the shortened CNTs. The adsorption level was monitored by UV spectroscopy (titration of unbound budesonide in the supernatant) which indicated that budesonide did not adsorb spontaneously on the nanotubes surface. This result highlights the central role of amphiphile 1 for the loading of the drug on CNT and its solubilization. |
| This journal is © The Royal Society of Chemistry 2016 |