DOI:
10.1039/C6RA09788J
(Paper)
RSC Adv., 2016,
6, 60407-60421
Synthesis, crystal structures, and in vitro anticancer properties of new N-heterocyclic carbene (NHC) silver(I)- and gold(I)/(III)-complexes: a rare example of silver(I)–NHC complex involved in redox transmetallation†
Received
15th April 2016
, Accepted 13th June 2016
First published on 13th June 2016
Abstract
A series of cationic, linearly coordinated silver(I)– (4–6) and gold(I)/(III)–NHC (NHC = N-heterocyclic carbene) (7–9) complexes of (benz)imidazol-2-ylidene ligands was prepared and successfully characterized. Complexes 4–6 were prepared by in situ deprotonation of azolium salts and 7–9 by NHC transfer from their respective silver(I) complexes. All new compounds were characterized by elemental analysis and 1H, 13C NMR, and FTIR spectroscopy techniques, and four of the complexes were unambiguously characterized by single-crystal X-ray diffraction method. Results of structural studies on single crystals of 5a, 6, and 8 revealed a linear coordination geometry about the metal center, whereas gold(III) in complex 7 had a consistent square-planar geometry. The reaction of complex 4 with gold(I) source formed a gold(III)–NHC complex following redox transmetallation. In all these cases, metal⋯H and π⋯π stacking interactions were observed, which affected the Ccarbene–M–Ccarbene bond angle. Preliminary anticancer activities of all new compounds were also studied in vitro against four human derived cancer cell lines by MTT assay method. Cationic silver(I) complexes displayed better anticancer potentials than their gold(I) counterparts. In particular, gold(III) complex displayed superior activity, with IC50 values at low-micro and nanomolar levels; thus, this complex showed superior selectivity toward all four tested cancer cell lines.
Introduction
Research on new transition metal N-heterocyclic carbene (NHC) complexes and their development for various applications have rapidly boomed over the past decade. A wide range of silver(I)- and, to a minor extent, gold(I)-complexes of both functionalized and nonfunctionalized NHCs have been investigated for their widespread applications in biology,1 catalysis,2 supramolecular chemistry,3 and photophysics.4 Considerable attention has also been given to the use of silver(I)–NHC complexes as carbene transfer candidates, in which NHCs can be transferred to a number of transition/inner transition metals. The use of these complexes as NHC transfer agents provides, in many cases, an appropriate way to overcome the practical difficulties caused by the use of inert atmospheres, strong bases, and problematic workups. In particular, the simpler synthetic methodologies developed for the preparation of catalytically active ruthenium(II)– and osmium(II)–NHC complexes for olefin metathesis5 and biologically active gold(I)–NHC complexes6 involve the technique of transmetallation from their respective silver(I) counterparts. Although, the use of silver complexes as carbene transfer agents is nowadays well developed, to the best of our knowledge, very little is known about their use as redox transmetallation agents. Arnold and Scarisbrick reported7 a binuclear alkoxy-bridged silver(I)–NHC complex employed in the preparation of ruthenium(III)–NHC complex, where the former functions as a redox transmetallating agent that undergoes reduction to form metallic silver, whereas ruthenium(II) is oxidized.
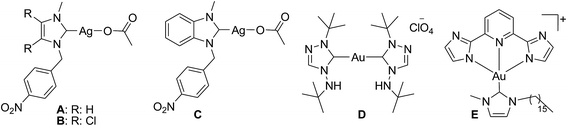
The design and development of group 10 and 11 metal complexes for anticancer applications8 have undergone a considerable growth as complements to platinum(II) compounds such as cisplatin, carboplatin, and oxaliplatin, among others, which are known as anticancer agents. Particularly, the four-coordinated square-planar cisplatin targets the DNA of the infected cell and intercalates into it to arrest the further cell propagation steps so as to kill the infected cell. Whereas, the gold(III) complexes of the similar ligand architecture targets the thioredoxin reductase (TrxR) for the inhibition of infected cell replication. In specific, the NHC–silver complex has earned a special place in the design of metal-based drugs as anticancer and antimicrobial agents in recent years,9 which probably stems from their low toxicity profile. To tune the possible anticancer potential of NHC–silver complexes, a variety of N-alkyl/aryl/functionalized NHCs that can induce both electronic and steric changes have been reported. With the (benz)imidazole-based silver(I)–NHC complexes showing exceptional performances as anticancer agents against a variety of cancer cell line (Chart 1: A, B and C),10 the need for expanding the scope of NHCs to those derived from other group 11 transition metal NHC complexes thus becomes obvious. Although the use of gold(I)/(III) complexes containing various mono- or bis-NHC ligands has been reported (D and E),11 their potential as anticancer agents have substantially been investigated.12 Complexes A, B and C found active against Caki-1 cells with the cytotoxicity of 20 ± 1, 17 ± 1 and 16 ± 9 μM, respectively, while the cationic gold(I) complex D evinced potent anticancer activity with IC50 value of 2.1 ± 0 μM against HT-29 and 1.0 ± 0 μM against MDA-MB-231. Interestingly, the square-planar gold(III)–NHC complex E displayed promising anticancer activity with an IC50 value of 1.4 ± 0.2 against HeLa cells. Much interest in the development of group 11 metal–NHC complexes as anticancer agents arises from the findings of a caffeine-derived silver(I)–NHC complex used as an antimicrobial agent under the trademark Silvamist®,13 a gold-based compound auranofin treated as an antirheumatic agent. Several other silver and gold complexes also show considerable potential against different cancer cells.14 To the best of our knowledge, only a handful of examples are available for benzimidazole-based gold(I)–NHC and gold(III)–NHC complexes employed for such purposes.
 |
| | Chart 1 Biologically potent group 11 metal–NHC complexes. | |
As our contribution to this particular area of research on transition metal–NHCs and their applications as metal-based drugs, we report herein the syntheses, structural characterization, and preliminary comparative anticancer potentials of a series of silver(I)– and gold(I)/(III)–NHC complexes having a (benz)imidazol-2-ylidene ligands. Furthermore, a rare example of a silver(I)–NHC complex involved in redox transmetallation to form gold(III)–NHC complex is presented.
Results and discussion
Synthesis of NHC proligands and complexes
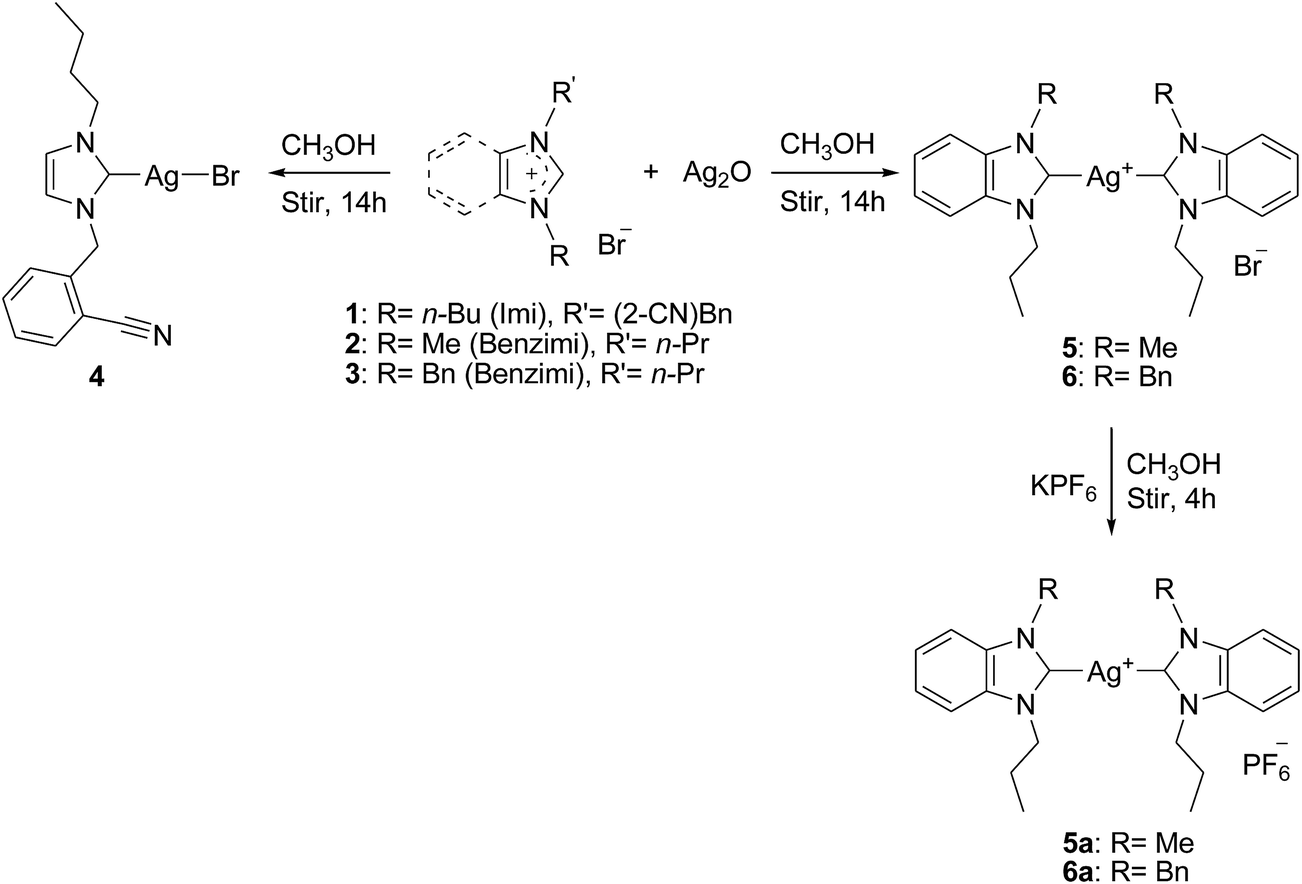
All the reported compounds were synthesized and characterized following procedures published earlier by our group15 and many others16 with slight modifications. Both functionalized and nonfunctionalized NHC proligands applied in this work were synthesized via the successive N-alkylation of (benz)imidazole with alkyl/aryl–alkyl bromides at basic condition. With the starting materials such as 1-n-butylimidazole and 1-methyl/1-benzylbenzimidazole synthesized, the ensuring quaternization reaction with 2-bromomethylbenzonitrile and n-propylbromide in refluxing acetonitrile for 20 h allowed for the isolation of NHC proligands 1–3, respectively, in good yield (Scheme 1). NHC–silver(I) complexes were prepared via the procedure reported by Wang and Lin.17 Mono- and bis-NHC silver(I) complexes 4–6 were prepared by the reaction of NHC proligands 1–3, respectively, with silver(I) oxide in methanol for 14 h at room temperature in the dark. Based on literature, this method of in situ deprotonation of azolium salts is widely used for the preparation of stable NHC–silver(I) complexes.18 The reactions involved in the preparation of NHC–silver(I) complexes are outlined in Scheme 2. Furthermore, for convenience in handling and increased crystalline nature, bromide–silver complexes 5 and 6 were treated with KPF6 in methanol to convert them into their hexafluorophosphate counterparts (Scheme 2). Latter complexes afforded their hexafluorophosphate–silver complexes 5a and 6a in good yield.
 |
| | Scheme 1 Preparation of carbene proligands 1–3. | |
 |
| | Scheme 2 Preparation of silver(I)–NHC bromide complexes 4–6 and silver(I)–NHC hexafluorophosphate complexes 5a and 6a. | |
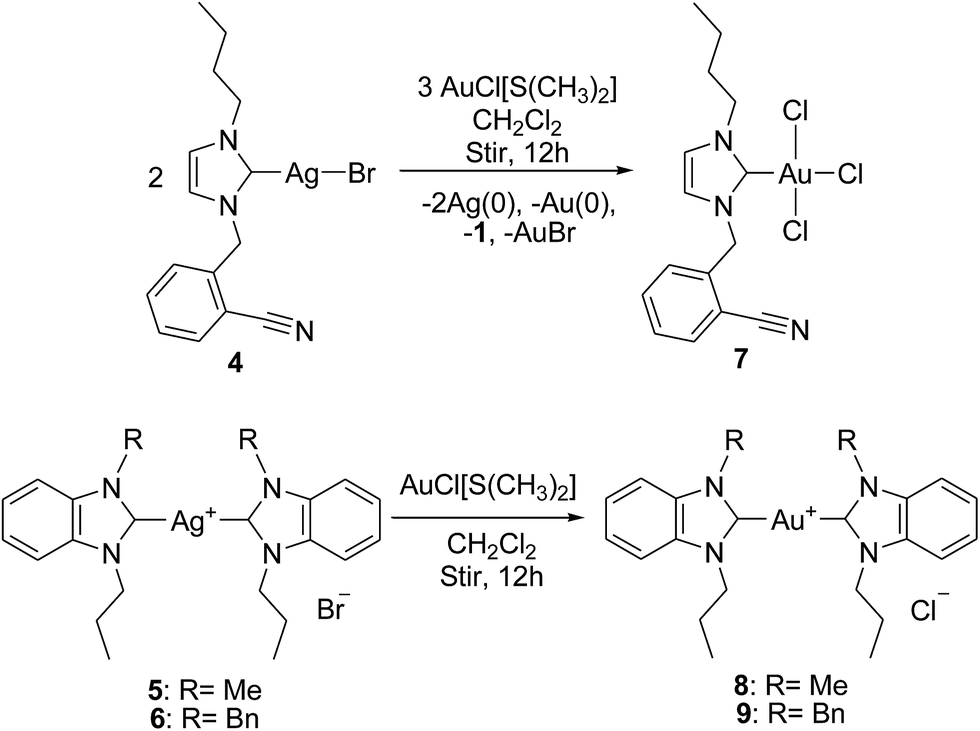
The use of new substituted mono- and bis-(benz)imidazol-2-lyidene silver(I) complexes as viable NHC transfer materials allowed for the preparation of gold(I)/(III) complexes 7–9 bound to the new (benz)imidazol-2-ylidene ligand system. More precisely, NHC–silver(I) complexes 4–6 were stirred with [AuCl(SMe)2] in dichloromethane for 12 h at room temperature in the dark to yield NHC–gold complexes 7–9, respectively, in comparable yield. As shown in Scheme 3, instead of gold(I), a tetra-coordinated gold(III) complex was formed following the redox transmetallation pathway. This procedure is probably one of the most commonly used techniques for the preparation of stable transition metal–NHC complexes at mild reaction conditions, thereby allowing an easy access to a library of compounds.19 Gold(III) complex was formed with a quantitative yield of almost 31.4%, whereas other gold complexes were formed in good yields. This particular reaction was performed in acetonitrile and again allowed for the formation of the same redox transmetallated product (gold(III) complex 7). This reaction allowed for the formation gold(III) complex by the assumption that the three chlorides originated from the Au(I) starting material and not from the chlorinated solvent. All three proligands and NHC–silver(I) complex 4 are brown/colorless viscous liquids, and silver complexes 5 and 6 and all gold complexes are stable solids at room temperature. All compounds are stable in atmospheric air and even with the presence of moisture (as indicated by NMR even after a week). All the reported compounds are soluble in polar organic solvents such as methanol, ethanol, acetonitrile, DMF and DMSO but insoluble in diethyl ether and hexane.
 |
| | Scheme 3 Transmetallation from silver(I)–NHC complexes 4–6 to gold(I)/(III)–NHC complexes 7–9. | |
NMR spectral characterization
All reported compounds were characterized by 1H and 13C{1H} NMR spectral techniques over the range 0–10 and 0–220 ppm, respectively. All compounds are stable in the solution upon exposure to air and moisture at room temperature for several days. In d6-DMSO, the 1H NMR resonance for the central (benz)imidazolium proton (C2H) of NHC proligands 1–3 is observed within the range 9.47–9.87 ppm, which is in accordance with those for the reported analogous compounds.20 The resonances for the inequivalent butyl, propyl, and backbone protons of the (benz)imidazole appear in the range 0.85–4.25, 0.95–4.48, and 7.40–8.13 ppm, respectively. Their 13C NMR spectra displayed resonances for alkyl and aryl carbon nuclei in the range 11.2–49.6 and 112–134 ppm, respectively, along with the central (benz)imidazolium carbon (C2) resonating at a more downfield region ca. 140 ppm, which is consistent with the formation of the desired azolium salts.21
The 1H NMR spectra of both silver(I) and gold(I)/(III) complexes show the expected ligand signals with no resonance peak for central (carbene) (benz)imidazolium proton, thereby indicating the deprotonation of the C2 proton and the coordination of carbene carbon to the metal center. The 13C NMR spectra of the NHC-complexes are more informative and conclusive because they show the resonance of the carbene carbon nuclei at the most downfield region in the range 177.7–189.5 for silver complexes and 171.3–184.9 ppm. Apart from these observations, the spectra show almost similar peaks for the resonance of other proton and carbon nuclei compared with their respective NHC proligands. The 13C carbene signals change most significantly, and a considerable upfield shift of ca. 10–37 ppm is noted upon the oxidation of Au(I) to Au(III) in the NHC complexes.22 In the case of NHC–Au(III) complex 7, the chemical shift value for the carbene carbon nucleus decreases by 9.4 ppm compared with the corresponding bromide–silver complex 4 because of the increased electron density caused by the presence of three chlorido-ligands around the metal, which in turn increase the Lewis acidity of the gold(III) center. This halido-depending or, in particular, chlorido-depending upfield shift caused by the shielding effect was also observed in a previous report by Huynh et al., in which 1,3-dialkyl-substituted benzimidazol-2-ylidenes were used as NHC ligands.23 Nevertheless, in the case of complexes 8 and 9, the Lewis acidity of the gold(I) centers is less than that of gold(III), thereby resulting in a downfield 13C carbene shifts of 184.9 and 178.3 ppm, respectively. These 13C carbene signals for the NHC–Au(I) complexes 8 and 9 well agree with our study24 and many other earlier reports.25 The chemical shifts for the carbene carbon atoms in NHC proligands and NHC-complexes are given in Table 1.
Table 1 Comparison of the diagnostic 13C carbene carbon chemical shift value of the NHC proligands and complexes
| Compound |
13CC2 (in δ) |
Δδa |
| Chemical shift difference value to the respective NHC proligands. Chemical shift difference value to the respective precursor Ag(I) complex. |
| Im-nBu-CN (1) |
138.2 |
— |
| Benzim-nPr-Me (2) |
139.3 |
— |
| Benzim-nPr-Bn (3) |
143.5 |
— |
| [AgIBr(Im-nBu-CN)] (4) |
180.7 |
42.5 |
| [AgI(Benzim-nPr-Me)2]Br (5) |
189.5 |
50.2 |
| [AgI(Benzim-nPr-Bn)2]Br (6) |
177.7 |
34.2 |
| [AgI(Benzim-nPr-Me)2]PF6 (5a) |
189.5 |
50.2 |
| [AgI(Benzim-nPr-Bn)2]PF6 (6a) |
177.7 |
34.2 |
| [AuIIICl3(Im-nBu-CN)] (7) |
171.3 |
33.1 |
| [AuI(Benzim-nPr-Me)2]Cl (8) |
184.9 |
45.6 (4.6)b |
| [AuI(Benzim-nPr-Bn)2]Cl (9) |
178.3 |
34.8 (0.6)b |
FTIR spectral characterization
The ability of pendant nitrile-functionality to coordinate with the metal center in a straightforward manner also gives access to a set of hemilabile complexes where NHCs function as chelating ligands.26 To know the possibility of nitrile coordination in the case of complexes 4 and 7 and the successful formation of the other desired compounds, FTIR spectral analysis was carried out. In the FTIR spectrum of proligand 1, a characteristic nitrile vibration band ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) with medium intensity is observed at ca. 2220 cm−1. This band remains unaltered in the complex spectra of 4 and 7, thereby indicating its noninvolvement in the coordination either with silver(I) or gold(III). Further, a remarkable decrease in the (benz)imidazole ring ν(C
N) with medium intensity is observed at ca. 2220 cm−1. This band remains unaltered in the complex spectra of 4 and 7, thereby indicating its noninvolvement in the coordination either with silver(I) or gold(III). Further, a remarkable decrease in the (benz)imidazole ring ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–N) absorption (ca. 1485 and 1080 cm−1) for all the complexes from the corresponding NHC proligands (ca. 1570 and 1175 cm−1) strongly suggests the formation of the desired carbene complexes. These absorption values and their assignments are in agreement with those for similar proligands and carbene complexes.27 The spectra of all reported compounds also show two distinguished bands of medium intensity at ca. 2860 and 2940 cm−1, which can be ascribed to the stretching mode of the vibration of aliphatic and aromatic C–H modules, respectively.
N and C–N) absorption (ca. 1485 and 1080 cm−1) for all the complexes from the corresponding NHC proligands (ca. 1570 and 1175 cm−1) strongly suggests the formation of the desired carbene complexes. These absorption values and their assignments are in agreement with those for similar proligands and carbene complexes.27 The spectra of all reported compounds also show two distinguished bands of medium intensity at ca. 2860 and 2940 cm−1, which can be ascribed to the stretching mode of the vibration of aliphatic and aromatic C–H modules, respectively.
Single crystal X-ray diffraction analysis of complexes
The results of the single crystal X-ray diffraction studies of the complexes for the structure determinations are consistent in terms of topology and stoichiometry, as depicted in the schemes. Suitable single crystals of silver complexes 5a and 6 and gold complexes 7 and 8 were subjected to X-ray diffraction analysis. The crystal data and structure refinement details for the complexes are provided in Table S1.† Pertinent bond distances and bond angles of complexes 5a, 6, 7, and 8 are tabulated in Tables 2–5, respectively.
Table 2 Selected bond distances (Å) and angles (°) for 5a
| Bond distance (Å) |
| Ag1–C1 |
2.083(2) |
N2–C2 |
1.394(3) |
| N1–C1 |
1.357(3) |
N1–C7 |
1.391(3) |
| N2–C1 |
1.357(3) |
C7–C2 |
1.398(3) |
| N1–C8 |
1.469(3) |
C2–C3 |
1.388(3) |
| N2–C11 |
1.464(3) |
C7–C6 |
1.393(3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Bond angle (°) |
| C1–Ag1–C1A |
180.0 |
C1–N2–C11 |
124.67(19) |
| N1–C1–Ag1 |
126.55(15) |
C1–N1–C7 |
110.84(17) |
| N2–C1–Ag1 |
127.31(15) |
C1–N2–C2 |
110.90(17) |
| N1–C1–N2 |
106.14(18) |
N2–C2–C7 |
105.86(18) |
| C1–N1–C8 |
123.87(18) |
N1–C7–C2 |
106.26(18) |
Table 3 Selected bond distances (Å) and angles (°) for 6
| Bond distance (Å) |
| Ag1–C1 |
2.205(14) |
N4–C18 |
1.37(3) |
| Ag1–C18 |
1.98(2) |
N1–C8 |
1.53(4) |
| N1–C1 |
1.24(2) |
N2–C15 |
1.46(2) |
| N2–C1 |
1.35(2) |
N3–C25 |
1.50(3) |
| N3–C18 |
1.37(3) |
N4–C32 |
1.44(3) |
|
| Bond angle (°) |
| C1–Ag1–C18 |
174.3(7) |
C18–N3–C25 |
122.8(18) |
| C1–N2–C15 |
124.6(15) |
C18–N4–C32 |
129(2) |
| C1–N1–C8 |
134(2) |
N3–C18–N4 |
105.1(19) |
| N1–C1–N2 |
115.9(15) |
N3–C25–C26 |
110(2) |
| N2–C1–Ag1 |
127.0(10) |
N4–C32–C33 |
122.1(19) |
Table 4 Selected bond distances (Å) and angles (°) for 7
| Bond distance (Å) |
| Au1–C10 |
1.994(10) |
N2–C10 |
1.343(12) |
| Au1–Cl1 |
2.338(2) |
N1–C11 |
1.466(15) |
| Au1–Cl2 |
2.3701(16) |
N2–C7 |
1.463(14) |
| Au1–Cl3 |
2.3816(14) |
N1–C9 |
1.386(14) |
| N1–C10 |
1.335(13) |
N2–C8 |
1.385(13) |
|
| Bond angle (°) |
| C10–Au1–Cl1 |
179.0(3) |
Au1–C10–N1 |
127.0(7) |
| Cl2–Au1–Cl3 |
176.70(6) |
Au1–C10–N2 |
126.2(7) |
| C10–Au1–Cl3 |
88.8(3) |
N1–C10–N2 |
106.8(8) |
| Cl3–Au1–Cl1 |
91.59(7) |
N1–C11–C12 |
122.7(11) |
| Cl1–Au1–Cl2 |
91.56(7) |
N2–C7–C6 |
111.7(9) |
| Cl2–Au1–C10 |
88.0(3) |
C10–N2–C7 |
125.1(9) |
Table 5 Selected bond distances (Å) and angles (°) for 8
| Bond distance (Å) |
| Au1–C1 |
2.028(9) |
N4–C12 |
1.349(12) |
| Au1–C12 |
2.028(9) |
N1–C8 |
1.471(12) |
| N1–C1 |
1.362(12) |
N2–C9 |
1.470(11) |
| N2–C1 |
1.329(11) |
N3–C19 |
1.450(11) |
| N3–C12 |
1.344(11) |
N4–C20 |
1.472(12) |
|
| Bond angle (°) |
| C1–Au1–C12 |
177.0(4) |
Au1–C12–N4 |
126.4(6) |
| N1–C1–N2 |
106.6(7) |
C1–N1–C8 |
126.7(8) |
| N3–C12–N4 |
107.1(8) |
C1–N2–C9 |
125.5(8) |
| Au1–C1–N1 |
121.7(6) |
C12–N3–C19 |
125.9(8) |
| Au1–C1–N2 |
131.6(7) |
C12–N4–C20 |
126.4(8) |
| Au1–C12–N3 |
126.5(7) |
N2–C9–C10 |
111.8(9) |
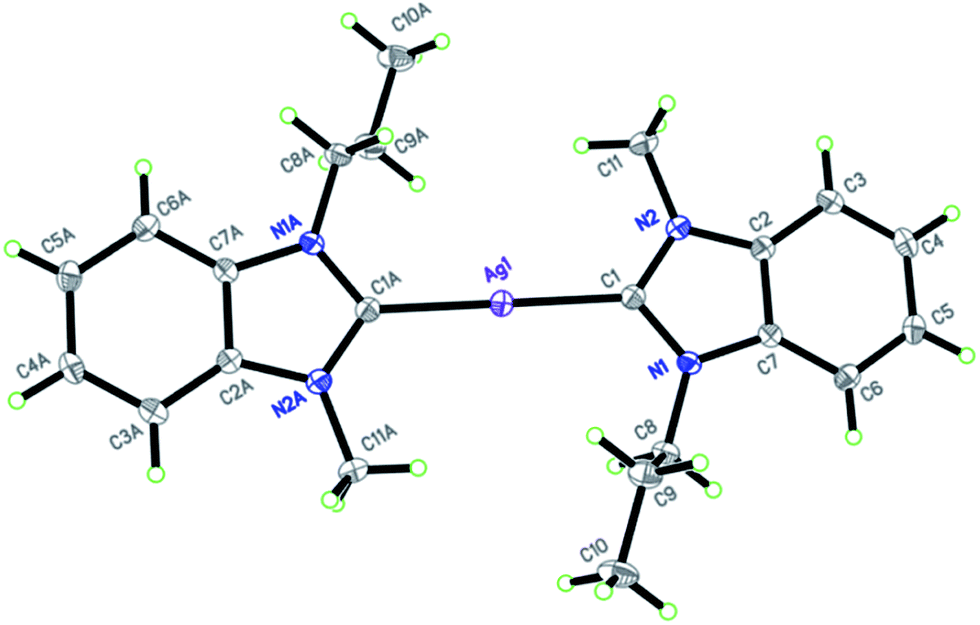
The molecular structure of complex 5a and 6 are shown in Fig. 1 and 2, respectively. Silver complex 5a crystallized in the monoclinic space group C2/c with one half of the complex cation and one half of the hexafluorophosphate anion occupied an asymmetric unit, whereas complex 6 crystallized in the triclinic space group P1 having one complete complex molecule in an asymmetric unit. The former has been defined as a well-ordered silver complex, whereas in the latter case, both the NHC ligands (syn-arrangement) are disordered over two sets with slightly different bond angles and distances. Ccarbene–Ag–Ccarbene in complex 5a is exactly linear with a bond angle of 180.0°, whereas in complex 6, the bond angle is 171.5(10)°, which is more distorted from linearity because of the close interaction of propyl-C1 protons with the silver center. The interaction bond distances are 2.685(5) and 2.933(3) Å, which are sufficiently shorter than the sum of the van der Waals radii of silver and hydrogen (2.92 Å). In complex 5a, propyl-C2 protons displayed close interaction with the metal center with an interaction bond distance of 2.972(5) Å on both sides of the silver center as NHC ligands possess anti-arrangement. The bond angle of silver(I) and the protons involved in a close interaction with the metal center, (C2)Hpropyl–Ag–(C2)Hpropyl, is exactly 180.0°. Thus, the Ccarbene–Ag–Ccarbene bond angle became linear despite of CH⋯Ag close interactions. The bond distance of silver(I)–Ccarbene modules and the internal ring angle of all benzimidazole rings at the carbene carbon center (N–C–N) range within 1.98(2)–2.205(14) Å and 105.1(19)–106.14(18)°, respectively. Furthermore, complex 6 possesses a weak π–π stacking interaction between the benzyl rings of NHC ligands with an interaction distance of 3.019 Å. The observed values for CH⋯silver(I) and π–π stacking interactions and other bond distances and angles are consistent with the values reported in a previous report.28 In the extended structure, complex cations are connected with the counter hexafluorophosphate/bromide anions into three-dimensional molecular arrays through C–H⋯F (2.535 Å) hydrogen bonds.
 |
| | Fig. 1 X-ray diffraction structure of silver(I) complex 5a with displacement ellipsoids drawn at 50% probability. Hexafluorophosphate anion was omitted for clarity. | |
 |
| | Fig. 2 X-ray diffraction structure of silver(I) complex 6 with displacement ellipsoids drawn at 50% probability. Bromide anion and acetonitrile molecule were omitted for clarity. | |
In the neutral gold(III) complex 7, one entire molecule comprises the asymmetric unit of the structure, which crystallized in the monoclinic space group P21/c. The molecular structure of complex 7 is depicted in Fig. 3. Complex 7 adopts an essentially square-planar geometry about the Au(III) center with slight deviations. The C10–Au–Cl1 and Cl2–Au–Cl3 bond angles are 179.0(3)° and 176.70(6)°, respectively, which agree well with data from literature.29 Similarly, Au–Ccarbene and Au–Cl bonds were in the typical range with bond distances of 1.994(10) and 2.338(2)–2.3816(14) Å, which are in agreement with the typical values for a gold(III)–NHC complex.30 The two chlorido ligands (Cl2 and Cl3) cis- to the NHC ligand deviated by about 4° from linearity, which is probably due to the interactions between the lone pairs of electrons of Cl2 and Cl3 with Cl1 ligand or with the empty p-orbital of the carbene carbon atom. This effect was also observed in similar trichloro-coordinated gold(III)–NHC complexes.31 The internal bond angle at the carbene carbon atom N1–C10–N2 is 106.8(8)°, which is shorter than the N–C–N bond angle in other imidazole based NHC proligands. The interatomic distances in the square-plane, namely, C10–Cl2, Cl2–Cl1, Cl1–Cl3, and Cl3–C10, are 3.044, 3.374, 3.383, and 3.075 Å, respectively, which are also within the sum of the van der Waals radii for carbon and chlorine (3.45 Å) and chlorine and chlorine (3.50 Å). The bond angle in the nitrile carbon is 177.0(14)°, which makes the bond almost linear. The nitrile nitrogen is pointing away from the gold center with a distance of 4.251 Å. Thus, any interaction between them is avoided. This observation is in agreement with the assignment based on FTIR spectroscopy results. In the crystal packing, hydrogen bonding interactions are observed between the benzylic protons and the chlorido ligand trans- to the carbene carbon with a bond distance of 2.764 Å along with other short contacts.
 |
| | Fig. 3 X-ray diffraction structure of gold(III) complex 7 with displacement ellipsoids drawn at 50% probability. | |
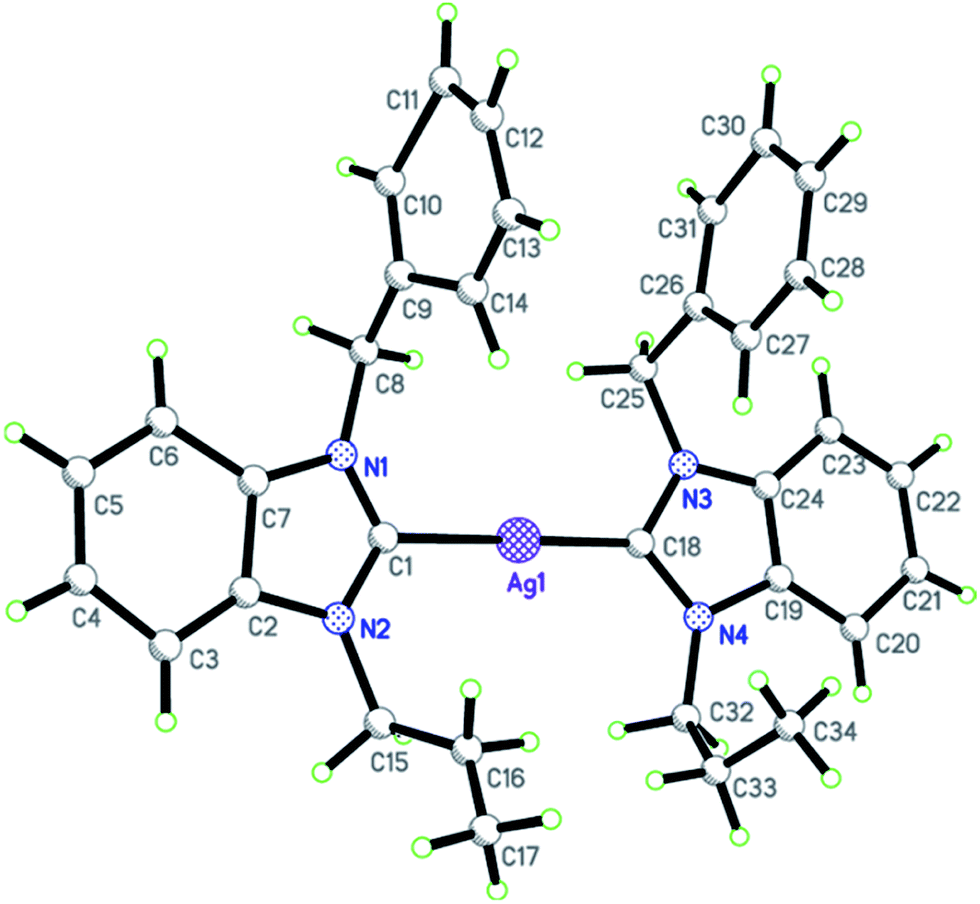
In the case of cationic gold(I) complex 8 (Fig. 4), the unit cell consists of one molecule of the complex cation, one chloride anion, and two crystal-held water molecules. Complex 8 is a well-ordered cationic bis-NHC–gold(I) complex crystallized in the monoclinic space group P21/c. Both the NHC ligands are positioned opposite to each other around the gold(I) center, thereby giving the complex a slightly distorted linear coordination geometry with the bond angle of 177.0(4)° for C1–Au–C12. The distances of Au–Ccarbene bonds, C1–Au and C12–Au, are being exactly same (2.028(9) Å). The internal bond angles at both the carbene carbon centers, N1–C1–N2 and N3–C12–N4, are found to be 106.6(7) and 107.1(8)°, respectively which are slightly greater than the typical values mentioned for the similar mono-/bis-NHC–gold(I) complexes.32,12a Furthermore, similar to its precursor silver(I)–NHC complex 5a, propyl C2 protons displayed an interaction with the gold(I) center with the interaction bond distance of 2.933 Å. However, the two NHC ligands in complex 8 are close to coplanarity (dihedral angle of 3.3(15)°), whereas those in complex 5a are coplanar. Interestingly, weak face-to-face π–π stacking interactions are observed between the two benzimidazole rings of adjacent molecules with an interaction distance of 3.563 Å,33 which is absent in the case of its silver(I) analog. In the extended structure, three types of intermolecular hydrogen bonds, namely, OH–Cl (3.173 Å), CH–Cl (2.905 Å), and CH–O (2.605 Å), are observed. These hydrogen bonds, along with other short interactions, collectively linked the complex cations and chloride anions into a three-dimensional network.
 |
| | Fig. 4 X-ray diffraction structure of gold(I) complex 8 with displacement ellipsoids drawn at 50% probability (A). Face-to-face π–π interactions between two adjacent molecules of 8 (B). Chloride anion and water molecules were omitted for clarity. | |
Redox transmetallation studies
In addition to providing a common transmetallation route to transition metal–NHC complexes, the redox transmetallation from silver(I)– or mercury(II)–NHC complexes to other transition metal–NHC complexes is also observed for particular alkoxide bridged systems.7,34 Silver(I)–NHC complexes are rarely used as carbene transfer agents for the preparation of other transition metal–NHC complexes via redox route where silver(I) is reduced to the metallic silver while the incoming metal is oxidized.7 Recently, Baker et al. reported34 a rare transmetallation reaction from a binuclear mercury(II)–NHC complex to palladium even in highly polar solvents such as DMSO at moderate temperature. These redox-transmetallation reactions allow the synthesis of a palladium(II)–NHC derivative, starting from the reaction of a mercury(II)–NHC complex with a palladium(0) source. Transmetallation involving silver(I)–NHC complexes is now a routine method for the synthesis of other transition metal–NHC derivatives, but this report on a redox transmetallation reaction involving a silver(I)–NHC complex is rare. In view of the apparently labile nature of the reported silver(I)–NHC complexes, their use in transmetallation to prepare gold(I)/(III)–NHC complexes is presented. A dicoordinated gold(I)–NHC complex was expected by the transmetallation reaction of complex 4 with a gold(I) source. However, as depicted in Scheme 3, silver complex 4 is treated with [AuCl(SMe)2] in dichloromethane for 12 h at room temperature and eventually resulted in the formation of a trichloro coordinated gold(III)–NHC complex 7 via redox transmetallation route. After 12 h of reaction, the reaction mixture consisted of a colorless solution and black precipitate (presumably metallic silver with traces of gold bromide and gold). The mixture was filtered and the solution was evaporated to dryness at reduced pressure to leave a pale yellow powder. Recrystallization of this product from dichloromethane/diethyl ether affords a microcrystalline compound characterized as the trivalent gold(III)–NHC complex. Further, the formation of gold(III)–NHC complex via redox transmetallation is supported by the yield of the complex, which is 31.4%. The reduction/oxidation of silver(I)/gold(I) to silver(0)/gold(III), however, only occurs for the neutral bromido-coordinated silver(I)-system 4 and not for the other systems at the same reaction conditions.
In vitro anticancer studies
The importance of silver-based anticancer agents has attracted considerable attention over the past decade.1 The ease of synthesis and functionalization of NHCs makes them promising ligand systems for the preparation of silver–NHC complexes and has encouraged researchers to use them in medicinal applications. Currently, interest in silver(I) and gold(I)/(III) complexes of NHCs containing different substitutions at 1,3-positions of both imidazole and benzimidazole core is increasing. These NHCs can endow the metal center with different steric environments by which resultant complexes can be tuned according to the target.
The positive results obtained from the anticancer screening of the reported silver(I)– and gold(I)–carbene complexes35 have encouraged us to test the cytotoxicity of carbene precursors (1–3) and their complexes (4–9) against human-derived colon cancer cell line, HCT 116 by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, in which mitochondrial dehydrogenase activity was measured as a sign of cell viability. In the present study, modifications made to the substituents at both the N-terminals of (benz)imidazole core allow for the independent tuning of the steric and electronic environment around silver(I) as well as gold(I) centers. This tuning certainly affects the lipophilicity of the test compounds leading to the definite action against cancer cells. A preliminary investigation of the anticancer potential of the reported compounds as a function of inhibition of cell proliferation was determined at different concentrations (0.01–100 μM). The anticancer capabilities of the active compounds to arrest the proliferation of HCT 116 cells at different concentrations are shown in Fig. 5A and B, whereas the IC50 values of all compounds are tabulated in Table 6. Similarly, the dose-dependent anti-proliferation activities of the Au–carbene complexes (7, 8, and 9) against human breast carcinoma (MCF-7), prostate (PC3), and leukemia (U937) cells are depicted in Fig. 6–8, respectively. Generally, cells from the negative control group showed fully confluent growth with a compact layer of proliferating HCT 116 cells. Cells treated with the standard drug 5-fluorouracil (IC50 = 5.2 ± 0.9 μM) showed decreased viability. NHC-proligand 1 and its silver complex 4 displayed almost no activity with IC50 values higher than 200 μM without affecting the normal morphology of the cells. For proligands 2 and 3, considerable anticancer activity was observed and affected the normal morphology of the cells and displayed decreased cell viability yielding IC50 values of 7.0 ± 0.8 and 57.5 ± 1.8 μM, respectively. Interestingly, benzimidazole-based silver(I)–NHC complexes 5 and 6 displayed moderate anticancer activities yielding IC50 values of 11.9 ± 0.9 and 19.9 ± 1.3 μM, respectively. This activity of the silver complexes can almost be compared with the analogues silver(I)acetate complexes A, B, and C against human renal cancer (Caki-1) cells and the bis-NHC silver complex D against human colorectal adenocarcinoma (HT-29) cells. However, it is difficult to correlate the structure with the activity of the complexes studied here in this report and in the cases of complexes A–D.
 |
| | Fig. 5 Dose-dependent cytotoxic activity against HCT 116 cells. (A) NHC-proligands (2 and 3), Ag– (5 and 6), and Au–carbene complexes (8 and 9); (B) Au–carbene complex 7. | |
Table 6 Anticancer potential of compounds 1–9 against HCT 116 cells expressed as IC50 values
| Compound |
IC50 (in μM) |
| Im-nBu-CN (1) |
>200 |
| Benzim-nPr-Me (2) |
7.0 ± 0.8 |
| Benzim-nPr-Bn (3) |
57.5 ± 1.8 |
| [AgIBr(Im-nBu-CN)] (4) |
>200 |
| [AgI(Benzim-nPr-Me)2]Br (5) |
11.9 ± 0.9 |
| [AgI(Benzim-nPr-Bn)2]Br (6) |
19.9 ± 1.3 |
| [AuIIICl3(Im-nBu-CN)] (7) |
0.05 ± 0.01 |
| [AuI(Benzim-nPr-Me)2]Cl (8) |
49.9 ± 2.4 |
| [AuI(Benzim-nPr-Bn)2]Cl (9) |
23.4 ± 1.8 |
| 5-Fluorouracil |
5.2 ± 0.9 |
 |
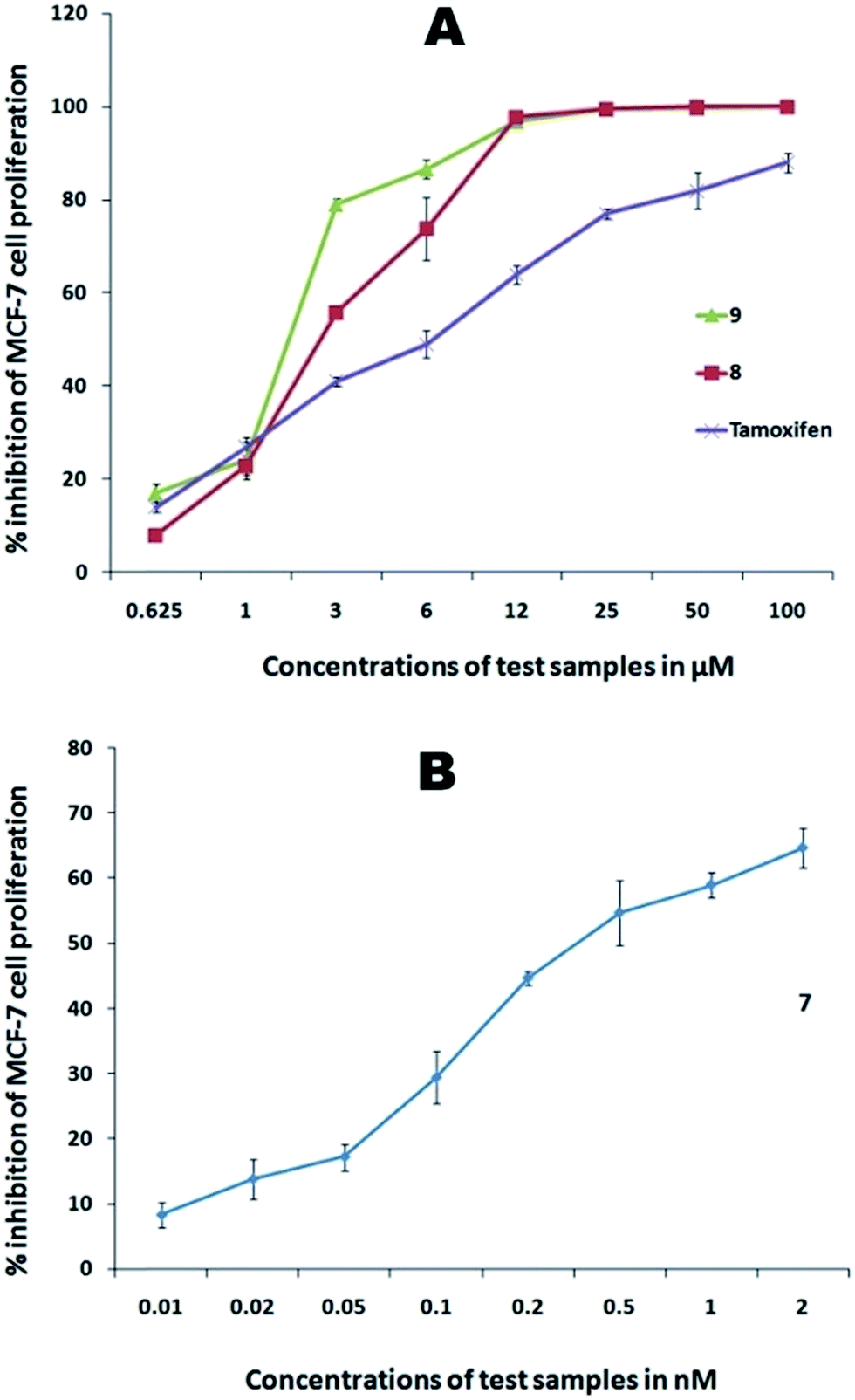
| | Fig. 6 Dose-dependent cytotoxic activity against MCF-7 cells. (A) Au–carbene complexes (8 and 9); (B) Au–carbene complex 7. | |
 |
| | Fig. 7 Dose-dependent cytotoxic activity against PC3 cells, (A) Au–carbene complexes (8 and 9); (B) Au–carbene complex-7. | |
 |
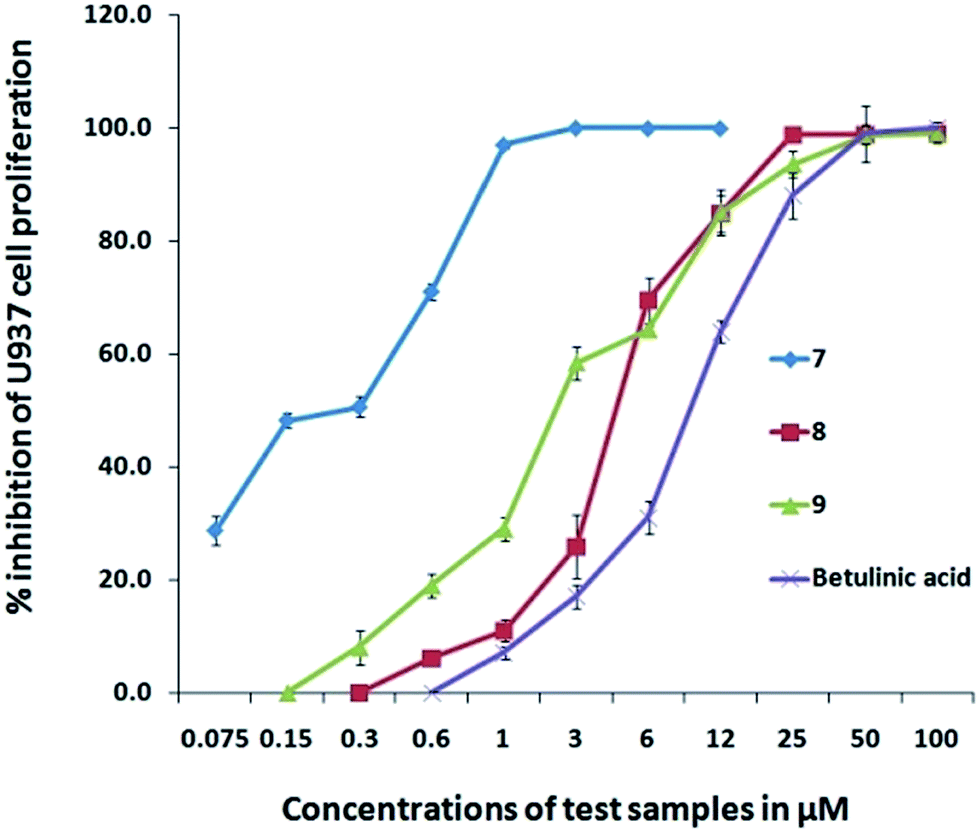
| | Fig. 8 Dose-dependent cytotoxic activity of Au–carbene complexes (8 and 9) and Au–carbene complex-7 against U937 cells. | |
After 72 h of incubation of the aforementioned concentrations, the gold(III) complex showed the highest activity among the tested compounds with an IC50 value of 0.05 ± 0.01 μM, which is 100 times more active than the standard used. This activity of the complex found more active than the reported gold(III)–NHC complex E (IC50: 1.4 ± 0.2 μM) against HeLa cells. Gold(I)–NHC complexes 8 and 9, however, displayed moderate anticancer activities against the tested cell line, yielding IC50 values of 49.9 ± 2.4 and 23.4 ± 1.8 μM, respectively, which are much weaker than that of the gold(III) complex. Among these complexes, silver(I) complexes showed better activity than their gold(I) counterparts. Gold(III) complex 4 showed promising anticancer activity compared with both silver(I) and gold(I) complexes. These findings could be unambiguously attributed to the steric and electronic effects of the carbene ligands as well as the nature of the metal ions that in turn depend on the lipophilicity of the test compounds.36 These observations are in agreement with previously reported activities of similar NHC complexes37 and delocalized lipophilic cations, which can readily enter into and accumulate in the mitochondria through the lipid membrane that result in mitochondria-induced apoptosis.38 This result is further confirmed by the photomicrographs, which reveal that the cells underwent severe characteristic morphological changes with nuclear condensation, and membrane blebbing can be seen clearly in Fig. 9. Furthermore, the characteristics in the HCT 116 cells treated with these complexes displayed an abnormally large number of vacuoles in the cytoplasm of the cancer cells. However, this prediction is only derived via indication, which may not be regarded as a sufficiently reliable probe because the anticancer potential of the test compounds also depends on other factors. The observed anticancer potentials of the carbene precursors and their complexes are comparable with those of structurally similar analogs.39
 |
| | Fig. 9 Photomicrographic images of HCT 116 cells treated with compounds 2 (100 μM), 3 (100 μM), 5 (100 μM), 6 (100 μM), 7 (0.15 μM), and 9 (100 μM) after 72 h of incubation. For the negative control, 0.1% DMSO was used, whereas 5-fluorouracil (100 μM) was used as the standard reference drug. | |
Much success has been achieved in terms of testing HCT 116 cells against gold(III)–NHC complex 7. This promising result prompted us to extend this study to other cancer cell lines such as human hormone-sensitive and invasive breast cancer (MCF-7), human prostate cancer (PC3), and human histiocytic leukemia (U937) cell lines by using gold complexes 7–9. Furthermore, their activities against human colorectal normal cell line (CCD-18Co) were also studied to investigate their selectivity indexes. For comparison purposes, the anticancer potentials of tamoxifen and betulinic acid 5-fluorouracil were evaluated at the same experimental conditions against MCF-7 and PC3, U937, and CCD-18Co, respectively. The IC50 values of these compounds against each cell line are tabulated in Table 7. The anticancer abilities of the active compounds to arrest the proliferation of the aforementioned cancer cells at different concentrations of the test compounds are shown in the ESI.†
Table 7 Anticancer potential of gold complexes 7–9 against MCF-7, PC3, U937, and CCD-18Co cells expressed as IC50 values
| Compound |
IC50 (in μM) |
| MCF-7 |
PC3 |
U937 |
CCD-18Co |
| 7 |
0.00031 ± 0.00002 |
0.00034 ± 0.00002 |
0.19 ± 0.002 |
17.7 ± 0.8 |
| 8 |
7.62 ± 0.4 |
126.27 ± 1.7 |
4.7 ± 0.4 |
135.2 ± 1.6 |
| 9 |
7.32 ± 0.8 |
5.58 ± 0.6 |
1.7 ± 0.06 |
71.2 ± 1.1 |
| Tamoxifen |
9.4 ± 0.4 |
— |
— |
— |
| Betulinic acid |
— |
17.0 ± 0.9 |
9.0 ± 0.7 |
6.7 ± 0.7 |
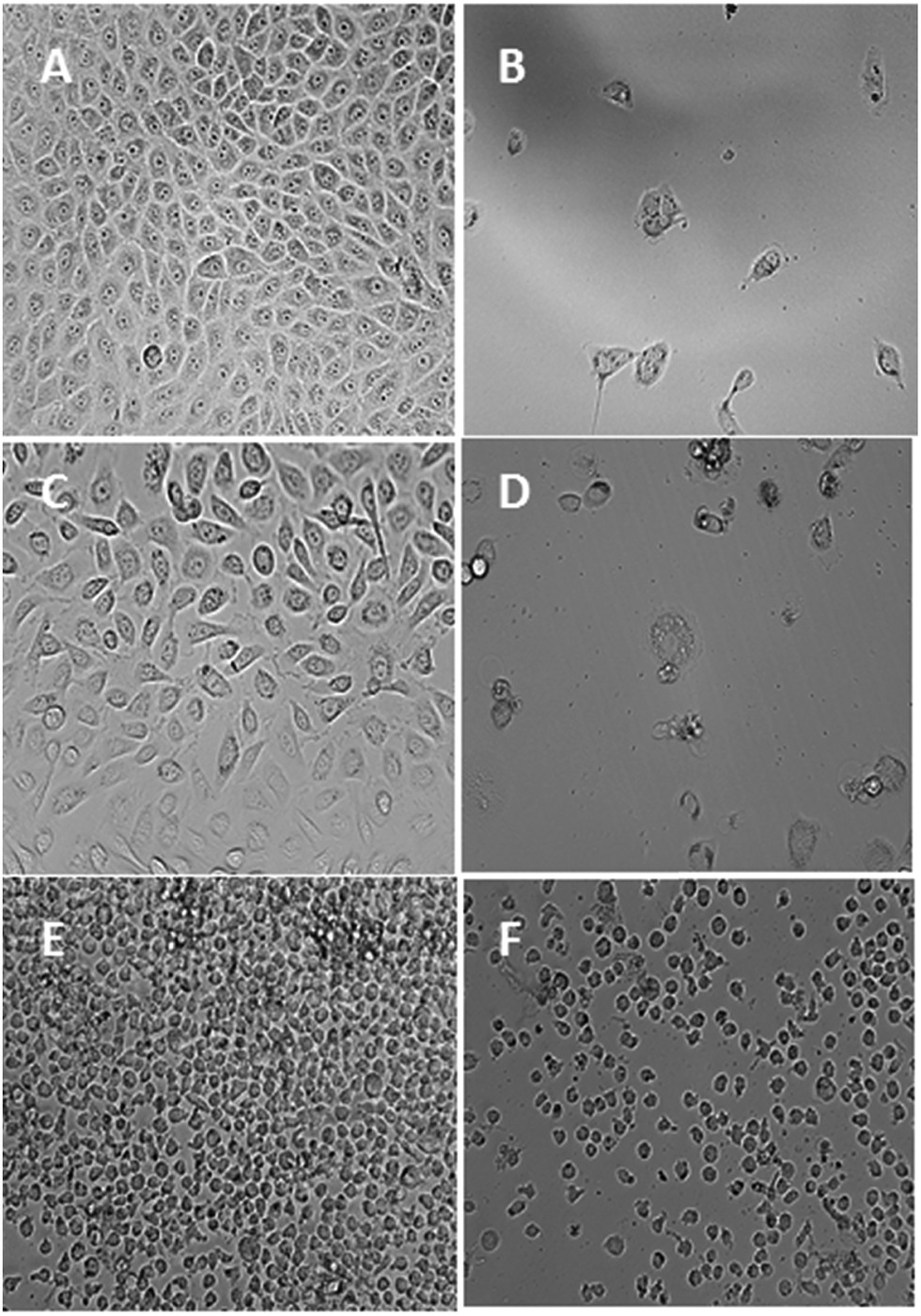
As shown in Table 7, gold(III) complex 7 presented high antiproliferative activity in the low-nanomolar level of 0.31 ± 0.02 and 0.34 ± 0.02 nM against MCF-7 and PC3 cell lines, respectively. Furthermore, its activity against the U937 cell line had an IC50 value of 0.19 ± 0.002 μM is nearly 20-fold lower than the standard used. On the other hand, gold(I) complex 8 displayed poor activity against PC3 cell line with an IC50 value of 126.27 ± 1.7 μM whereas complex 9 presented a much better activity in the micromolar level of 5.58 ± 0.6 μM. However, complexes 8 and 9 were very active against MCF-7 and U937 with IC50 values of 7.62 ± 0.4, 7.32 ± 0.8, and 4.7 ± 0.4, and 1.7 ± 0.06 μM, respectively, which are less than the IC50 values of the standards measured at the same experimental conditions. From a chemical point of view, the analysis of these data show that the gold(III) complex demonstrated a remarkably higher anticancer potential against all the four cancer cell lines, thereby indicating that this system is very suitable in terms of anticancer activity. This result could be possibly caused by their redox activities, which can occur at the cellular environment. Thus, one oxidation state of the metal center becomes stable against another. Further, the aforementioned complex is more labile compared with the other complexes, and any biological activation of this type of complexes via ligand detachment is more likely favorable because it bears three labile chloride ligands. Complex 7 affected the normal morphology of almost all the tested cancer cells (Fig. 10). Except for a few dead cells, only debris were observed in the cell pictures. Similarly, other gold complexes also affected the normal morphology of the cancer cells and showed marked signs of cytotoxicity caused by the changes in the viable characteristic features. Certainly, further insights into the activation and mode of action of the complexes are essential to better appreciate the significance of the relative activities of the tested gold complexes. A plausible mechanism of gold(III) complex against MCF-7 and PC3 cell lines is likely dominated by the grouping of the gold-based inhibition of thioredoxin reductase (TrxR) and antimitochondrial effects related to the neutral nature of the complex, while in other two cases, HCT 116 and U937 cell lines, the DNA of the cancer cell may be the target. Later mode of action of the planar complex might have yielded in the lesser anticancer activity compared with TrxR inhibition.10,11 Since, perhaps the targets of attack of this complex are different, hence the variation in the corresponding anticancer potential was observed though there is no much difference is reported between these cell lines in the DTP NCI 60 cell line panel. It is not possible to arrive at a conclusion with this preliminary data, and more studies necessary to establish the mode of action and the difference in the anticancer potential against similar cancer cell lines.
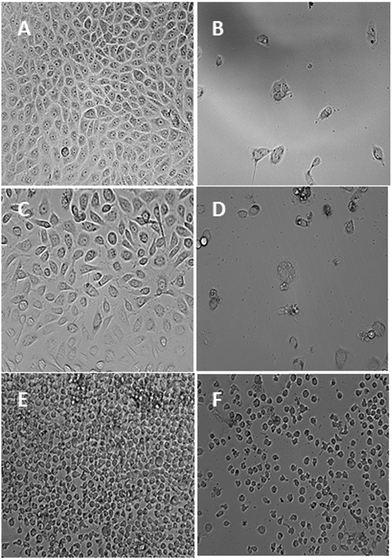 |
| | Fig. 10 Photomicrographic images of MCF-7 cells representing the negative control (A) and the complex-7 ((2.0 μM)) treated cells (B). Photomicrographic images of PC3 cells representing the negative control (C) and the complex-7 (2.0 μM) treated cells (D). Photomicrographic images of U937 cells representing the negative control (E) and the complex-7 (12.0 μM) treated cells (F). All the images were taken after 72 h of incubation at 200× magnification. | |
Selectivity index studies
Although the cytotoxic effects of the tested compounds toward many cancer cell lines are promising, these candidates should not harm normal cells. The selectivity index, which indicates the cytotoxic selectivity of the compound against cancer cells and its safety toward the normal cells, was determined from the ratio of the IC50 value obtained from the test on normal cell (CCD-18Co) versus the IC50 value for cancer cells (HCT 116, MCF-7, PC3, and U937). The sample with a selectivity index value higher than 3 is suggested to have a high selectivity toward a particular cell line. To determine the reactivity of gold complexes toward normal cells, complexes 7–9 were treated with human colorectal normal cell line (CCD-18Co) cells at the aforementioned experimental conditions. All the gold complexes were found to be less toxic against normal cell line and in general displayed activities that are 2–20 orders of magnitude lower than that of the standard. However, useful mono-cultures of CCD-18Co cells have been and continue to be in research and industry to compare the activity of a new drug molecule against cancer and non-cancer cells, it cannot be ignored that, the higher concentration of drug is required to inhibit CCD-18Co cells probably in relation with its higher doubling time in culture. This makes the cells resistant to cytotoxic agents, nevertheless, this study was under observation for its higher doubling time once in every 3 h, and no such doubling is found under the mentioned experimental conditions. Similar reports are found in the literature,40 which can be compared with the present study. The selectivity index values of gold complexes 7–9 for the tested cancer cell lines are shown in Table 8. This analysis revealed that the neutral gold(III) complex 7 demonstrated a remarkably higher selectivity toward all the four cancer cell lines compared with other compounds.
Table 8 Selectivity index of the gold complexes for representative cancer cell lines
| Compound |
Selectivity indexa |
| HCT 116 |
MCF-7 |
PC3 |
U937 |
| Selective index = IC50 on normal (CCD-18Co) cell line/IC50 on cancer cell line. |
| 7 |
354.0 |
57096.0 |
52059.0 |
93.1 |
| 8 |
2.7 |
17.7 |
1.1 |
28.7 |
| 9 |
3.0 |
9.7 |
12.7 |
41.8 |
Conclusions
We reported a series of silver(I) and gold(I)/(III)–NHC complexes with varying steric bulks around the metal center, type of NHC ligand, and metal atoms. Silver and gold complexes were accessed in a straightforward manner via in situ deprotonation of azolium salts and NHC transfer protocols, respectively. All compounds were characterized by spectral and analytical techniques. Interestingly, a square-planar gold(III)–NHC complex is formed via redox transmetallation route where silver(I) undergoes reduction to form metallic silver, whereas gold(I) forms gold(III) upon oxidation. In the crystal structures of the complexes, metal–H interactions were observed, which affect the Ccarbene–metal–Ccarbene module. A preliminary anticancer study of all nine compounds against HCT 116 cancer cell line revealed the promising anticancer potential of gold(III)–NHC complex 4 with an IC50 value of 0.05 μM. On the other hand, silver(I) complexes displayed better activities in comparison to their gold(I) counterparts. This difference in their performance depends on the relative lipophilicity of cationic silver(I)– and gold(I)–NHC complexes in the cellular environment, thereby favoring the slow release of silver ions because silver–C bonds are more labile than gold–C bonds. From the photomicrographs, the test compounds react with the HCT 116 cells that result in mitochondria-induced apoptosis, as confirmed by the occurrence of nuclear condensation and membrane blebbing. Furthermore, gold complexes displayed promising activity against the MCF-7, PC3, and U937 cell lines. Gold(III) complex 7 presented IC50 values in nanomolar concentration level against former two cell lines and considerable activity against the latter. Selectivity index results revealed that complex 7 had a higher selectivity toward all four cancer cell lines compared with other compounds. We also plan to investigate the exact mode of action and mechanism of these complexes against all four tested cancer cell lines and other cell lines in the future.
Experimental section
General considerations
All chemicals and solvents were obtained from commercial sources and used as received without further purifications. 1-Butylimidazole, 1-methylbenzimidazole, benzimidazole, benzyl bromide, 1-propyl bromide, 2-bromomethylbenzonitrile, silver(I)oxide, [AuCl(SMe)2], potassium hexafluorophosphate, 5-fluorouracil (standard used in anticancer activity), and 3-[4,5-yl]-2,5-diphenyltetrazolium bromide used for the MTT assay were purchased from Sigma-Aldrich. NMR spectra were recorded either in d6-DMSO or CDCl3 by using a Bruker 500 MHz spectrometer. Chemical shifts (δ) are given in ppm with TMS as the internal reference. Signals are labeled as singlet (s), doublet (d), triplet (t), and multiplet (m). The melting points were assessed using a Stuart Scientific (UK) instrument. Elemental analysis was carried out using a PerkinElmer microanalyzer. The FT-IR spectra of the compounds were recorded in potassium bromide disks by using a Perkin Elmer 2000 system spectrometer over the range 4000 to 400 cm−1. The instruments are available at The School of Chemical Sciences, Universiti Sains Malaysia (USM). The X-ray diffraction data were collected using a Bruker CCD area-detector diffractometer. Calculations, structure refinement, molecular graphic design, and publishing were performed using the SHELXTL and PLATON software packages available at The School of Physics, USM.
Syntheses
Synthesis of 3-(benzonitrile-2-methyl)-1-butylimidazolium bromide (1). 2-Bromomethylbenzonitrile (0.390 g, 2 mmol) was added to a solution of 1-butylimidazole (0.248 g, 2 mmol) in acetonitrile (20 mL), and the resulting mixture was stirred for 20 h at 100 °C. The resulting solution was cooled to room temperature. The solvent was removed using a rotary evaporator to obtain the brown viscous oil of 1, which was then washed with diethyl ether (3 × 3 mL) and stored in a vacuum desiccator. Yield: 71.3%. 1H NMR (500 MHz, d6-DMSO, 298 K): δ 0.85 (3H, t, J = 6.5 Hz, CH3), 1.21 (6H, m, CH2–CH2–CH3), 1.75 (5H, m, CH2–CH2–CH3), 4.25 (2H, t, J = 7.0 Hz, N–CH2), 5.72 (2H, s, N–CH2Ar), 7.55, 7.65, 7.78 (4H, m, Ar-H), 7.95 (1H, s, imidazolium H4′), 7.98 (1H, s, imidazolium H5′), and 9.47 (1H, s, imidazolium H2′). 13C{1H} NMR (125 MHz, d6-DMSO, 298 K): δ 13.0 (CH3), 19.5 (CH2–CH2–CH3), 31.8 (CH2–CH2–CH3), 49.6 (N–CH2), 51.2 (N–CH2Ar), 117.6 (CN), 123.0, 125.2 (imidazolium-C4′ and C5′), 128.5, 130.5, 134.5, 137.6 (Ar-C), and 138.2 (imidazolium C2′). FT-IR (in cm−1): ∼2860, ∼2925 cm−1 ν(C–H aliphatic and aromatic), 2220 ν(CN), 1592, 1176 ν(imidazole ring CN, C–N).
Synthesis of 1-methyl-3-propylbenzimidazolium bromide (2). This compound was prepared in a manner analogous to that of 1, with 1-methylbenzimidazole (0.264 g, 2 mmol) and propyl bromide (0.244 g, 2 mmol) used instead of 1-butylimidazole and 2-bromomethylbenzonitrile. Yield: 74.5%. 1H NMR (500 MHz, d6-DMSO, 298 K): δ 0.95 (3H, t, J = 7.0 Hz, CH2–CH3), 1.93 (2H, m, CH2–CH3), 4.09 (3H, s, N–CH3), 4.48 (2H, t, J = 6.5 Hz, N–CH2), 7.75, 8.05, 8.13 (4H, m, Ar-H), and 9.85 (1H, s, benzimidazolium H2′). 13C{1H} NMR (125 MHz, d6-DMSO, 298 K): δ 11.2 (CH3), 23.6 (CH2–CH3), 35.5 (N–CH3), 49.9 (N–CH2), 111.9, 112.08, 121.4, 124.0 (Ar-C), 139.3 (benzimidazolium C2′). FT-IR (in cm−1): 2856, ∼2930 ν(C–H aliphatic and aromatic), 1588, 1190 ν(benzimidazole ring CN, C–N).
Synthesis of 1-benzyl-3-propylbenzimidazolium bromide (3). This compound was prepared in a manner analogous to that of 1, with 1-benzylbenzimidazole (0.416 g, 2 mmol) and propyl bromide (0.244 g, 2 mmol) used instead of 1-butylimidazole and 2-bromomethylbenzonitrile. Yield: 79.8%. 1H NMR (500 MHz, d6-DMSO, 298 K): δ 0.94 (3H, t, J = 6.5 Hz, CH2–CH3), 1.93 (2H, m, CH2–CH3), 4.48 (2H, t, J = 6.0 Hz, N–CH2Pr), 5.78 (2H, s, N–CH2) 7.40, 7.52, 7.68 (5H, m, benzyl-H), 7.96, 8.12 (4H, d, J = 7.6 Hz, benzimidazolium-H), and 9.97 (1H, s, benzimidazolium H2′). 13C{1H} NMR (125 MHz, d6-DMSO, 298 K): δ 10.6 (CH3), 21.9 (CH2–CH3), 48 (N–CH2Pr), 50.1 (N–CH2), 110.6, 113.8, 121.5, 124.7 (benzimidazolium-C), 128.5, 131.3, 133.7, 136.9 (benzyl-C) and 143.5 (benzimidazolium C2′). FT-IR (in cm−1): ∼2860, 2920 ν(C–H aliphatic and aromatic), 1582, 1196 ν(benzimidazole ring CN, C–N).
Synthesis of 3-(benzonitrile-2-methyl)-1-butylimidazoliumsilver(I)bromide (4). A suspension of 3-(benzonitrile-2-methyl)-1-butylimidazolium bromide 1 (0.319 g, 1 mmol) and silver(I)oxide (0.463 g, 2 mmol) in methanol (25 mL) was stirred at room temperature for 14 h in the dark. The reaction mixture was filtered through a pad of celite to remove unreacted silver(I)oxide. The solvent was then removed using a rotary evaporator to afford product 4 as a brown viscous oil. Yield: 70.2%. 1H NMR (500 MHz, CDCl3, 298 K): δ 0.93 (3H, t, J = 6.5 Hz, CH3), 1.34 (6H, m, CH2–CH3), 1.80 (5H, m, CH2–CH2–CH3), 4.14 (2H, t, J = 6.0 Hz, N–CH2), 5.52 (2H, s, N–CH2Ar), 7.08 (1H, d, J = 7.5 Hz, imidazolium H5′),7.16 (1H, d, J = 7.5 Hz, imidazolium H4′), and 7.50, 7.69 (4H, m, Ar-H). 13C{1H} NMR (125 MHz, CDCl3, 298 K): δ 13.1 (CH3), 19.5 (CH3–CH2–CH2), 33.1 (CH2–CH2–CH3), 52.2 (N–CH2), 53.5 (N–CH2Ar), 117.2 (CN) 122.1, 129.4 (imidazolium-C4′ and C5′) 133.5, 133.9, 139.5 (Ar-C), and 180.7 (Ccarbene–Ag). FT-IR (in cm−1): ∼2870, 2945 ν(C–H aliphatic and aromatic), 1482, 1064 ν(imidazole ring CN, C–N).
Synthesis of bis-(1-methyl-3-propylbenzimidazolium)silver(I)bromide (5). This complex was prepared in a manner analogous to that of 4, with 2 (0.255 g, 1 mmol) used instead of 1 to afford an off-white solid of 5. Yield: 93.2%, mp: 244–246 °C. 1H NMR (500 MHz, d6-DMSO, 298 K): δ 0.96 (3H, t, J = 6.0 Hz, CH3), 1.95 (2H, m, CH2–CH3), 4.15 (2H, s, N–CH3), 4.55 (2H, t, J = 5.5 Hz, N–CH2Pr), 7.50, 7.84, 7.91 (4H, m, benzimidazolium-H). 13C{1H} NMR (125 MHz, d6-DMSO, 298 K): δ 12.0 (CH3), 24.5 (CH2–CH3), 36.4 (N–CH3), 50.8 (N–CH2Pr), 111.9, 119.6, 124.4, 134.4, 146.8 (benzimidazolium-C), and 189.5 (Ccarbene–Ag). FT-IR (KBr disc) cm−1: ∼2860, 2933 ν(C–H aliphatic and aromatic), 1488, 1093 ν(benzimidazole ring CN, C–N). Anal. calc. for C22H28N4AgBr: C 49.3, H 5.3, N 10.4%. Found: C 48.9, H 5.5, N 10.3%.
Synthesis of bis-(1-benzyl-3-propylbenzimidazolium)silver(I)bromide (6). This complex was prepared in a manner analogous to that of 4, with 3 (0.330 g, 1 mmol) used instead of 1 to afford an off-white solid of 6. Yield: 82.2%, mp: 228–230 °C. 1H NMR (500 MHz, d6-DMSO, 298 K): δ 0.96 (3H, t, J = 6.0 Hz, CH3), 1.97 (2H, m, CH2–CH3), 4.45 (2H, t, J = 6.5 Hz, N–CH2Pr), 5.70 (2H, s, N–CH2), 7.19, 7.30, 7.38 (4H, m, benzimidazolium-H), and 7.85 (3H, d, J = 7.0 Hz, Ar-H), 7.96 (2H, t, J = 6.5 Hz, Ar-H). 13C{1H} NMR (125 MHz, d6-DMSO, 298 K): δ 11.5 (CH3), 23.7 (CH2–CH3), 48.8 (N–CH2Pr), 51.0 (N–CH2), 110.0, 120.4, 126.9 (benzimidazolium-C), 129.2, 134.1, 143.5 (Ar-C), and 177.7 (Ccarbene–Ag). FT-IR (KBr disc) cm−1: 2869, 2937 ν(C–H aliphatic and aromatic), 1496, 1088 ν(benzimidazole ring CN, C–N). Anal. calc. for C34H36N4AgBr·CH3CN: C 59.3, H 5.4, N 9.6%. Found: C 59.1, H 5.6, N 9.0%.
Salt metathesis of silver(I)–NHC bromide complexes (5 and 6) to silver(I)–NHC hexafluorophosphate complexes (5a and 6a). A solution of silver(I)–NHC complex 5 (0.300 g, 0.5 mmol) in methanol (20 mL) was added KPF6 (0.092 g, 0.5 mmol) and stirred for 3–4 h at room temperature. The solvent was removed using a rotary evaporator to afford a white solid of 5a. This residual solid was thoroughly washed with water to remove unreacted KPF6 and was isolated by filtration and dried. In the same manner, silver bromide complex 6 was successfully converted into its hexafluorophosphate counterpart 6a. Furthermore, the NMR and FTIR spectral data of the hexafluorophosphate derivatives are as same as their bromide derivatives. The elemental analysis results of these complexes are in agreement with their structure. Single crystals suitable for X-ray diffraction studies were grown by the diffusion of diethyl ether into the acetonitrile solution of the complex.
Synthesis of trichlorido-[3-(benzonitrile-2-methyl)-1-butylimidazolium]gold(III) (7). A mixture of silver–NHC complex 4 (0.053 g, 0.125 mmol) and [AuCl(SMe)2] (0.045 g, 0.125 mmol) in dichloromethane (20 mL) was stirred at room temperature for 12 h. The formation of a black precipitate (presumably metallic silver with trances gold bromide and gold) was observed. This reaction mixture was filtered through a pad of celite and the solvent was removed using a rotary evaporator to yield product 7 as an off-white solid. Single crystals suitable for X-ray diffraction studies were grown by the diffusion of diethyl ether into the acetonitrile solution of the complex. Yield: 31.4%, mp: 246–248 °C. 1H NMR (500 MHz, CDCl3, 298 K): δ 0.87 (3H, t, J = 6.0 Hz, CH3), 1.30 (6H, m, CH2–CH2–CH3), 1.78 (5H, m, CH2–CH2–CH3), 4.13 (2H, t, J = 6.0 Hz, N–CH2), 5.50 (2H, s, CH2), 6.95 (1H, s, imidazolium H4′), 7.10 (1H, s, imidazolium H5′) and 7.44, 7.59, 7.68 (4H, m, Ar-H). 13C{1H} NMR (125 MHz, CDCl3, 298 K): δ 13.1 (CH3), 19.5 (CH2–CH2–CH3), 33.0 (CH2–CH2–CH3), 51.5 (N–CH2), 53.1 (N–CH2Ar), 117.3 (CN), 121.1, 129.5 (imidazolium C4′ and C5′), 133.2, 134.1, 138.5 (Ar-C), and 171.3 (Ccarbene–Au). FT-IR (KBr disc) cm−1: 2830, 2954 ν(C–H aliphatic and aromatic), 1474, ∼1070 ν(imidazole ring CN, C–N). Anal. calc. for C15H17N3AuCl3: C 33.2, H 3.2, N 7.7%. Found: C 33.3, H 3.5, N 7.5%.
Synthesis of bis-(1-methyl-3-propylbenzimidazolium)gold(I)chloride (8). This complex was prepared in a manner analogous to that of 7, with 5 (0.067 g, 0.125 mmol) used instead of 4 to afford an off-white solid of 8. Single crystals suitable for X-ray diffraction studies were grown by the diffusion of diethyl ether into the acetonitrile solution of the complex. Yield: 79.8%, mp: 217–219 °C. 1H NMR (500 MHz, d6-DMSO, 298 K): δ 0.98 (3H, t, J = 5.5 Hz, CH3), 2.00 (2H, m, CH2–CH3), 4.18 (2H, s, N–CH3), 4.62 (2H, t, J = 5.5 Hz, N–CH2Pr), 7.54, 7.87, 7.94 (4H, m, benzimidazolium-H). 13C{1H} NMR (125 MHz, d6-DMSO, 298 K): δ 15.7 (CH3), 18.6 (CH2–CH3), 37.4 (N–CH3), 45.1 (N–CH2Pr), 111.3, 120.1, 124.4, 128.8, 139.6 (benzimidazolium-C) and 184.9 (Ccarbene–Au). FT-IR (KBr disc) cm−1: 2832, 2956 ν(C–H aliphatic and aromatic), 1479, ∼1075 ν(imidazole ring CN, C–N). Anal. calc. for C22H28N4AuCl·2H2O: C 42.8, H 5.2, N 9.1%. Found: C 42.6, H 5.3, N 9.3%.
Synthesis of bis-(1-benzyl-3-propylbenzimidazolium)gold(I)bromide (9). This complex was prepared in a manner analogous to that of 7, with 6 (0.086 g, 0.125 mmol) used instead of 4 to afford an off-white solid of 9. Yield: 77.7%, mp: 225–227 °C. 1H NMR (500 MHz, d6-DMSO, 298 K): δ 1.00 (3H, t, J = 5.5 Hz, CH3), 1.99 (2H, m, CH2–CH3), 4.48 (2H, t, J = 5.0 Hz, N–CH2), 5.72 (2H, s, N–CH2), 7.17–7.48 (4H, m, benzimidazolium-H), 7.74 (2H, d, J = 6.5 Hz, Ar-H), 7.84, 7.98 (2H, t, J = 5.5 Hz, Ar-H). 13C{1H} NMR (125 MHz, d6-DMSO, 298 K): δ 11.4 (CH3), 23.3 (CH2–CH3), 48.8 (N–CH2), 52.8 (N–CH2Ar), 110.0, 120.4, 126.9 (benzimidazolium-C), 129.2, 134.1, 135.5 (Ar-C), and 178.3 (Ccarbene–Au). FT-IR (KBr disc) cm−1: 2868, 2923 ν(C–H aliphatic and aromatic), 1484, ∼1075 ν(imidazole ring CN, C–N). Anal. calc. for C34H36N4AuCl: C 55.7, H 5.0, N 7.6%. Found: C 55.4, H 5.3, N 7.6%.
Determination of anticancer activity
Cell culture. Initially, HCT 116, MCF-7, PC3, U937, and CCD-18Co cells were allowed to grow at optimal incubator conditions. Cells that have reached a confluence of 70–80% were chosen for cell plating purposes. The old medium was carefully aspirated out of the plate. The cells were then washed 2–3 times by using sterile phosphate buffered saline (PBS) with (pH 7.4). PBS was completely discarded after washing and trypsin was then added and distributed evenly onto the cell surfaces. The cells were incubated at 37 °C in 5% CO2 for 1 min. The flasks containing the cells were then gently tapped to facilitate cell segregation and were then observed using an inverted microscope (if cell segregation is not satisfactory, the cells were incubated for another minute). Trypsin activity was inhibited by adding 5 mL of fresh complete media of 10% fetal bovine serum (FBS). The cells were counted and diluted to obtain a final concentration of 2.5 × 105 cells per mL and inoculated into wells (100 μL cells per well). Finally, plates containing the cells were incubated at 37 °C with an internal atmosphere of 5% CO2.
Test sample preparation for bioassay. The compounds were first dissolved in DMSO to obtain the stock solutions of 10 mM concentration. Further, for biological assay, the stock solutions of the compounds were serially diluted using respective cultured media to obtain various concentrations (0.02, 0.05, 0.1, 0.2, 0.4, 0.78, 1.5, 3, 6, 12.5, 25, 50, 100, and 200 μM) of the test samples (working standards). From the prepared working standard, 100 μL was added to each well containing 100 μL of cells. Therefore, upon addition of the working standard into a well, each concentration was reduced to half of its original concentration. Accordingly, the final concentrations tested for all the compounds were 0.01, 0.02, 0.05, 0.1, 0.2, 0.4, 0.78, 1.5, 3, 6, 12.5, 25, 50, and 100 μM. Given that DMSO was used as to dissolve the compounds, 0.1% DMSO was used as the negative control.
MTT assay. Cancer cells (100 μL cells per well, 1.5 × 105 cells per mL) were inoculated in wells of microtitre plate. The plate was incubated in a CO2 incubator overnight to allow the cells to attach. About 100 μL of the test complexes were added into each well containing the cells. The test complexes were diluted with media into the desired concentrations from the stock. The plates were incubated at 37 °C with an internal atmosphere of 5% CO2 for 72 h. About 20 μL of MTT reagent was added into each well and incubated again for 4 h. About 50 μL of MTT lysis solution (DMSO) was added into the wells. The plates were further incubated for 5 min in the CO2 incubator. Finally, the plates were read at 570 and 620 nm wavelengths by using a standard ELISA microplate reader (Ascent Multiskan). Data were recorded and analyzed for the assessment of the effects of test complexes on cell viability and growth inhibition. The percentage of growth inhibition was calculated from the optical density (OD) that was obtained from MTT assay. The formula used for the calculation of growth inhibition was eqn (1). Tamoxifen and 5-fluorouracil were used as standard reference drugs for MCF-7 and HCT 116 cells, respectively. Betulinic acid was used as the positive control for U937, PC3, and CCD-18Co cells for comparison.| |
 | (1) |
Acknowledgements
R. A. H. thanks Universiti Sains Malaysia (USM) for the Research University (RU) grant 1001/PKIMIA/811217. M. Z. G. thanks USM for the RU grant 1001/PKIMIA/844137 and Kufa University, Najaf, Iraq for a postgraduate research scholarship.
References
-
(a) S. Budagumpi, R. A. Haque, S. Endud, G. U. Rehman and A. W. Salman, Eur. J. Inorg. Chem., 2013, 4367 CrossRef CAS
 ;
(b) A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884 CrossRef CAS ;
(c) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859 CrossRef CAS PubMed ;
(d) A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23 RSC ;
(e) W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755 RSC .
;
(b) A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884 CrossRef CAS ;
(c) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859 CrossRef CAS PubMed ;
(d) A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23 RSC ;
(e) W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755 RSC . -
(a) Y. He, M. F. Lv and C. Cai, Dalton Trans., 2012, 41, 12428 RSC ;
(b) A. S. K. Hashmi, Top. Organomet. Chem., 2013, 44, 143 CrossRef ;
(c) M. K. Samantaray, C. Dash, M. M. Shaikh, K. Pang, R. J. Butcher and P. Ghosh, Inorg. Chem., 2011, 50, 1840 CrossRef CAS PubMed .
-
(a) F. E. Hahn, C. Radloff, T. Pape and A. Hepp, Chem.–Eur. J., 2008, 14, 10900 CrossRef CAS PubMed ;
(b) A. Rit, T. Pape, A. Hepp and F. E. Hahn, Organometallics, 2011, 30, 334 CrossRef CAS .
-
(a) V. J. Catalano and A. L. Moore, Inorg. Chem., 2005, 44, 6558 CrossRef CAS PubMed ;
(b) S. D. Adhikary, S. K. Seth, M. R. Senapati and J. Dinda, J. Mol. Struct., 2013, 1042, 123 CrossRef ;
(c) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed .
-
(a) C. A. Urbina-Blanco, A. Poater, T. Lebl, S. Manzini, A. M. Z. Slawin, L. Cavallo, S. P. Nolan and J. Am, Chem. Soc., 2013, 135, 7073 CrossRef CAS PubMed ;
(b) A. K. Chatterjee, J. P. Morgan, M. Scholl and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 3783 CrossRef CAS ;
(c) T. Weskamp, F. J. Kohl, W. Hieringer, D. Gleich and W. A. Herrmann, Angew. Chem., Int. Ed., 1999, 38, 2416 CrossRef CAS ;
(d) R. Castarlenas, M. A. Esteruelas and E. Oñate, Organometallics, 2005, 24, 4343 CrossRef CAS .
-
(a) M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 3708 RSC ;
(b) M.-L. Teyssot, A.-S. Jarrousse, M. Manin, A. Chevry, S. Roche, F. Norre, C. Beaudoin, L. Morel, D. Boyer, R. Mahiou and A. Gautier, Dalton Trans., 2009, 6894 RSC ;
(c) A. John and P. Ghosh, Dalton Trans., 2010, 39, 7183 RSC .
- P. L. Arnold and A. C. Scarisbrick, Organometallics, 2004, 23, 2519 CrossRef CAS .
- R. A. Haque, A. W. Salman, S. Budagumpi, A. A. Abdullah and A. M. S. Abdul Majid, Metallomics, 2013, 5, 760 RSC .
-
(a) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med.
Chem., 2011, 54, 3 CrossRef CAS PubMed ;
(b) W. J. Youngs, A. R. Knapp, P. O. Wagers and C. A. Tessier, Dalton Trans., 2012, 41, 327 RSC .
-
(a) S. Patil, A. Deally, B. Gleeson, F. Hackenberg, H. Müller-Bunz, F. Paradisi and M. Tacke, Z. Anorg. Allg. Chem., 2011, 637, 386 CrossRef CAS ;
(b) S. Patil, A. Deally, B. Gleeson, H. Müller-Bunz, F. Paradisi and M. Tacke, Appl. Organomet. Chem., 2010, 24, 781 CrossRef CAS ;
(c) S. Patil, J. Claffey, A. Deally, M. Hogan, B. Gleeson, L. M. Menéndez Méndez, H. Müller-Bunz, F. Paradisi and M. Tacke, Eur. J. Inorg. Chem., 2010, 1020 CrossRef CAS .
-
(a) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC ;
(b) G. Ung, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 758 CrossRef CAS PubMed ;
(c) T. V. Serebryanskaya, A. A. Zolotarev and I. Ott, MedChemComm, 2015, 6, 1186 RSC ;
(d) T. Zou, C. T. Lum, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930 CrossRef CAS PubMed .
-
(a) H. Sivaram, J. Tan and H. V. Huynh, Organometallics, 2012, 31, 5875 CrossRef CAS ;
(b) C. Hartinger, C. N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677 CrossRef CAS .
-
(a) C. L. Cannon, L. A. Hogue, R. K. Vajravelu, G. H. Capps, A. Ibricevic, K. M. Hindi, A. Kascatan-Nebioglu, M. J. Walter, S. L. Brody and W. J. Youngs, Antimicrob. Agents Chemother., 2009, 53, 3285 CrossRef CAS PubMed ;
(b) W. J. Youngs, A. R. Knapp, P. O. Wagers and C. A. Tessier, Dalton Trans., 2012, 41, 327 RSC .
-
(a) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3 CrossRef CAS PubMed ;
(b) S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550 CAS ;
(c) P. J. Barnard and S. J. Berners-Price, Coord. Chem. Rev., 2007, 251, 1889 CrossRef CAS ;
(d) I. Ott, Coord. Chem. Rev., 2009, 253, 1670 CrossRef CAS .
-
(a) R. A. Haque, A. W. Salman, S. Budagumpi, A. A. Abdullah, Z. A. Abdul Hameed Al-Mudaris and A. M. S. Abdul Majid, Appl. Organomet. Chem., 2013, 27, 465 CrossRef CAS ;
(b) A. W. Salman, R. A. Haque, S. Budagumpi and H. Z. Zulikha, Polyhedron, 2013, 49, 200 CrossRef CAS .
-
(a) P. J. Barnard, L. E. Wedlock, M. V. Baker, S. J. Berners-Price, D. A. Joyce, B. W. Skelton and J. H. Steer, Angew. Chem., Int. Ed., 2006, 45, 5966 CrossRef CAS PubMed ;
(b) J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570 CrossRef CAS PubMed ;
(c) L. Kaps, B. Biersack, H. Müller-Bunz, K. Mahal, J. Münzner, M. Tacke, T. Mueller and R. Schobert, J. Inorg. Biochem., 2012, 106, 52 CrossRef CAS PubMed .
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972 CrossRef CAS .
- I. J. B. Lin and C. S. Vasam, Comments Inorg. Chem., 2004, 25, 75 CrossRef CAS .
-
(a) S. P. Nolan, Acc. Chem. Res., 2011, 44, 91 CrossRef CAS PubMed ;
(b) S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778 CrossRef CAS PubMed ;
(c) H. G. Raubenheimer and S. Cronje, Chem. Soc. Rev., 2008, 37, 1998 RSC .
- M. Z. Ghdhayeb, R. A. Haque and S. Budagumpi, J. Organomet. Chem., 2014, 757, 42 CrossRef CAS .
- R. A. Haque, S. Budagumpi, S. Y. Choo, M. K. Choong, B. E. Lokesh and K. Sudesh, Appl. Organomet. Chem., 2012, 26, 689 CrossRef CAS .
-
(a) H. G. Raubenheimer, P. J. Oliver, L. Lindeque, M. Desmet, J. Hrušak and G. J. Kruger, J. Organomet. Chem., 1997, 544, 91 CrossRef CAS ;
(b) R. Jothibasu, H. V. Huynh and L. L. Koh, J. Organomet. Chem., 2008, 693, 374 CrossRef CAS ;
(c) H. G. Raubenheimer and S. Cronje, J. Organomet. Chem., 2001, 617–618, 170 CrossRef CAS .
- H. V. Huynh, S. Guo and W. Wu, Organometallics, 2013, 32, 4591 CrossRef CAS .
- R. A. Haque, M. Z. Ghdhayeb, S. Budagumpi, A. W. Salman, M. B. Khadeer Ahamed and A. M. S. Abdul Majid, Inorg. Chim. Acta, 2013, 394, 519 CrossRef CAS .
-
(a) M. V. Baker, P. J. Barnard, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2005, 37 RSC ;
(b) X. Hu, I. Castro-Rodriguez, K. Olsen and K. Meyer, Organometallics, 2004, 23, 755 CrossRef CAS ;
(c) P. J. Barnard, M. V. Baker, S. J. Berners-Price, B. W. Skelton and A. H. White, Dalton Trans., 2004, 1038 RSC .
- H. Z. Zulikha, R. A. Haque, S. Budagumpi and A. M. S. Abdul Majid, Inorg. Chim. Acta, 2014, 411, 40 CrossRef .
- R. A. Haque, N. Hasanudin, M. A. Iqbal, A. Ahmad, S. Hashim, A. M. S. Abdul Majid and M. B. Khadeer Ahamed, J. Coord. Chem., 2013, 66, 3211 CrossRef CAS .
-
(a) S. Hameury, P. de Frémont, P. R. Breuil, H. Olivier-Bourbigou and P. Braunstein, Dalton Trans., 2014, 43, 4700 RSC ;
(b) Q.-X. Liu, H. Wang, X.-J. Zhao, Z.-Q. Yao, Z.-Q. Wang, A.-H. Chen and X.-G. Wang, CrystEngComm, 2012, 14, 5330 RSC ;
(c) R. A. Haque, A. W. Salman, C. K. Whai, C. K. Quah and H.-K. Fun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m97 CrossRef CAS PubMed .
- C. D. Abernethy, G. M. Codd, M. D. Spicer and M. K. Taylor, J. Am. Chem. Soc., 2003, 125, 1128 CrossRef CAS PubMed .
-
(a) T. Zou, C. T. Lum, S. S. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930 CrossRef CAS PubMed ;
(b) B. Jacques, J. Kirsch, P. de Frémont and P. Braunstein, Organometallics, 2012, 31, 4654 CrossRef CAS ;
(c) E. Tomas-Mendivil, P. Y. Toullec, J. Borge, S. Conejero, V. Michelet and V. Cadierno, ACS Catal., 2013, 3, 3086 CrossRef CAS .
- P. Shukla, J. A. Johnson, D. Vidovic, A. H. Cowley and C. D. Abernethy, Chem. Commun., 2004, 360 RSC .
-
(a) M. Bouhrara, E. Jeanneau, L. Veyre, C. Copereta and C. Thieuleux, Dalton Trans., 2011, 40, 2995 RSC ;
(b) F. Hackenberg, H. Müller-Bunz, R. Smith, W. Streciwilk, X. Zhu and M. Tacke, Organometallics, 2013, 32, 5551 CrossRef CAS ;
(c) R. Jothibasu and H. V. Huynh, Chem. Commun., 2010, 46, 2986 RSC .
- L.-A. Schaper, X. Wei, S. J. Hock, A. Pöthig, K. Öfele, M. Cokoja, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 32, 3376 CrossRef CAS .
- M. V. Baker, D. H. Brown, R. A. Haque, B. W. Skelton and A. H. White, J. Inclusion Phenom. Macrocyclic Chem., 2009, 65, 97 CrossRef CAS .
-
(a) R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646 CrossRef CAS PubMed ;
(b) W. Streciwilk, J. Cassidy, F. Hackenberg, H. Müller-Bunz, F. Paradisi and M. Tacke, J. Organomet. Chem., 2014, 749, 88 CrossRef CAS .
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608 CrossRef CAS PubMed .
-
(a) P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642 CrossRef CAS PubMed ;
(b) M. M. Jellicoe, S. J. Nichols, B. A. Callus, M. V. Baker, P. J. Barnard, S. J. Berners-Price, J. Whelan, G. C. Yeoh and A. Filipovska, Carcinogenesis, 2008, 29, 1124 CrossRef CAS PubMed ;
(c) A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692 CrossRef CAS .
-
(a) L. Ronconi, D. Aldinucci, Q. P. Dou and D. Fregona, Anti-Cancer Agents Med. Chem., 2010, 10, 283 CrossRef CAS ;
(b) M. P. Rigobello, G. Scutari, R. Boscolo and A. Bindoli, Br. J. Pharmacol., 2002, 136, 1162 CrossRef CAS PubMed .
-
(a) R. A. Haque, M. Z. Ghdhayeb, A. W. Salman, S. Budagumpi, M. B. Khadeer Ahamed and A. M. S. Abdul Majid, Inorg. Chem. Commun., 2012, 22, 113 CrossRef CAS ;
(b) M. A. Iqbal, R. A. Haque, S. Budagumpi, M. B. Khadeer Ahamed and A. M. S. Abdul Majid, Inorg. Chem. Commun., 2013, 28, 64 CrossRef CAS .
- M. L. Henriksson, S. Edin, A. M. Dahlin, P.-A. Oldenborg, Å. Öberg, B. V. Guelpen, J. Rutegård, R. Stenling and R. Palmqvist, Am. J. Pathol., 2011, 178, 1387 CrossRef PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1457027–1457030 contain the supplementary crystallographic data for the complexes 5a, 6, 7, and 8, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra09788j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.