DOI:
10.1039/C6RA09660C
(Paper)
RSC Adv., 2016,
6, 41594-41607
Dendrimer like star polymer based on β-cyclodextrin with ABC type miktoarms†
Received
14th April 2016
, Accepted 16th April 2016
First published on 20th April 2016
Abstract
The present article reports the synthesis and self aggregation study of a novel dendrimer like star polymer based on β-cyclodextrin (β-CD), in which the primary alcoholic arms of β-CD have been linked to ABC type miktoarm star polymers. Initially, 6-heptaazido-6-deoxy-β-cyclodextrin (7N3-β-CD) has been clicked with alkyne terminated poly(methyl methacrylate) (alkynyl-PMMA-Br), which has been synthesized through atom transfer radical polymerization technique. Subsequently, azidation has been done in order to prepare β-CD(PMMA-N3)7. Parallelly, an amphiphilic block copolymer (alkynyl-PDLL-b-PNIPAAm), having an alkyne end group, has been synthesized by employing a mikto functional initiator having alcohol, xanthate and alkyne functionalities, in ring opening polymerization (ROP) of D,L-lactide (DLL) from –OH end, and reversible addition-fragmentation chain-transfer (RAFT) polymerization of N-isopropylacrylamide (NIPAAm) from xanthate end. Then, the desired star polymer has been synthesized by the click reaction between β-CD(PMMA-N3)7 and alkynyl-PDLL-b-PNIPAAm. The polymers have been characterized by 1H NMR, and gel permeation chromatography. Self aggregation of the star polymers has been investigated by 1H NMR and fluorescence spectroscopy. Size of the micelles formed has been analyzed by dynamic light scattering and transmission electron microscopic techniques.
Introduction
In recent years, amphiphilic star shaped block copolymers like miktoarm star polymers have received much attention in terms of their synthesis and self assembly.1,2 Miktoarm star polymer is a star polymer which contains two or more arm species with different chemical compositions and/or molecular weights.3–6 The micelles formed by the miktoarm star polymers can provide distinct chemical environments to store various kinds of drug molecules, gene therapy agents, microreactors and capsulates.7–10 For the preparation of miktoarm star polymers, recently, many methods like combination of controlled/living radical polymerization routes and modular synthesis have been developed.11–14 Controlled/living radical polymerization methods involve ring opening polymerization (ROP), atom transfer radical polymerization (ATRP), reversible addition fragmentation chain transfer polymerization (RAFT) and nitroxide mediated radical polymerization (NMP)15,16 where as modular approach to chemical functionalization covers a class of chemical modifications, termed click chemistry, 1,3-dipolar cycloaddition and Diels–Alder reaction.17–21
Because of controlled/living radical polymerization methods synthesis of polymers with predetermined chemical composition and complex architectures has been significantly simplified. Moreover, these methods are applicable to a variety of monomers and have significant tolerance to experimental conditions in comparison with living ionic polymerization techniques.22 When these techniques are combined with modular approaches such as click reaction,23 star and branched polymers with extended compositions such as 3-arm ABC,24 4-arm ABCD star copolymers25 and ABCDE-type H-shaped quinto polymers26,27 have been successfully obtained.
Gungor et al. have synthesized a mikto functional initiator, propargyl-3-[(2-bromo-2-methylpropanoyl)oxy]-2-(hydroxymethyl)-2-methylpropanoate, and employed it in the synthesis of H-shaped (ABCDE type) quintopolymer.28 By using same initiator recently Ozcan Altintas et al. have reported dendrimer like miktoarm star terpolymers (A)3–(B–C)3 using a combination of click chemistry and multiple controlled/living polymerization methods such as ROP, NMP, and ATRP.29 Gou et al. has synthesized an amphiphilic drug conjugated A14B7 miktoarm star copolymer based on β-cyclodextrin, via the combination of controlled ring opening polymerization and click reaction.30
In this article, we have discussed the synthesis of a new mikto functional initiator having alcohol, xanthate and alkyne end groups, and its handling in synthesizing a novel dendrimer like star polymer based on β-cyclodextrin, in which all the primary alcoholic arms of β-cyclodextrin core are linked to ABC type miktoarm star polymers. Further the self assembly property of micelle formation, of the synthesized polymer has been studied. For the synthesis purpose, we have combined controlled/living radical polymerization methods (ATRP, ROP and RAFT) with modular approach (click reaction). The monomers employed in ATRP, ROP and RAFT, are methyl methacrylate (MMA), D,L-lactide (DLL) and N-isopropylacrylamide respectively. PMMA is a promising polymer for applications in optical, pneumatic actuation, sensor, analytical separation, and conductive devices. It is also useful in biomedical applications, polymer electrolytes, polymer viscosity, and drug delivery using electro-diffusion or electro-osmotic flow. Due to its compatibility and easy processing as a polymer moiety, PMMA with carbon nanotubes or other inorganic materials plays an important role in the development of nanotechnology.31 D,L-Lactide is one of the most commonly used monomers in ROP and the corresponding polymer has many biomedical applications. ROP of D,L-lactide has been done at room temperature by using an organo catalyst, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).32 ATRP of MMA has been done in general manner, using Cu(I)Br as a catalyst.33 Poly(N-isopropylacrylamide) (PNIPAM) is most widely studied thermo-responsive polymer due to its interesting thermal behavior. It undergoes volume phase transition (from coil to globular state) in water at around 33 °C. This temperature is known as its lower critical solution temperature (LCST).34 These interesting elements of these polymers made them to be chosen in order to synthesize the desired star polymer.
Experimental section
Materials
β-Cyclodextrin hydrate (β-CD) was purchased from Aldrich and dried for 48 h in vacuum oven before use. Propargyl alcohol (98%) (AVRA), sodium azide (NaN3) (99.5%, SDFCL), triethylamine (TEA) (SDFCL), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (99%, Sigma Aldrich), dry tetrahydrofuran (THF) (99.5%, Spectrochem), N,N-dimethylformamide (DMF) (99.5%, SDFCL) were used as received. 2-Bromo-2-methylpropanoyl bromide was purchased from Sigma Aldrich. But-3-yn-1-ol (98%) was purchased from Spectrochem, India. Copper(I) bromide (CuBr) (98% Himedia) was washed consecutively with glacial acetic acid, ethanol and diethyl ether, and then dried at 40 °C for 3 days in vacuum oven, and was stored under nitrogen atmosphere.35 Dry dichloromethane (DCM) (≥99%) was purchased from MERCK and distilled over CaH2 under nitrogen atmosphere. Azobis(isobutyronitrile) (AIBN) (Spectrochem, Mumbai, India, 98%) was recrystallized from methanol. Tetrahydrofuran (THF) (Spectrochem, Mumbai, India, ≥99%) was dried and fractionally distilled from sodium and benzophenone. DBU was purchased from Spectrochem and was used as received. Dialysis membrane 70, LA 393, Himedia, Mumbai; MWCO 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–14000 daltons; pore size: 2.4 nm was obtained.
000–14000 daltons; pore size: 2.4 nm was obtained.
Methyl methacrylate purchased from SD fine chemicals was washed with 5% NaOH solution, and then with distilled water. Later it was dried over CaH2 and then distilled under low pressure. DLL was purchased from Sigma Aldrich and was recrystallized in ethyl acetate. NIPAAm was purchased from Sigma Aldrich and was recrystallized in hexane just before use. Potassium O-ethylxanthate was prepared as reported in the literature.36 Heptakis(6-azido-6-deoxy)-β-cyclodextrin (7N3-β-CD) (1),37,38 propargyl-3-[(2-bromo-2-methylpropanoyl)oxy]-2-(hydroxymethyl)-2-methylpropanoate (2)28 and potassium O-ethyl carbonodithioate (3) were prepared according to the methods reported in the literature.
General methods
1H NMR spectra were recorded on a JEOL AL300 FTNMR (300 MHz) at room temperature and reported in parts per million (δ) from internal standard tetramethylsilane or residual solvent peak. The number average molecular weight (Mn,GPC) and polydispersity index (Mw/Mn) were determined by Youglin ACME 9000 gel permeation chromatography in DMF at 40 °C with flow rate 0.5 mL min−1 on two polystyrene gel columns (PL gel 5 μm 10 × 104 Å columns (300 × 7.5 mm)) connected in series to Younglin ACME 9000 Gradient Pump and a Younglin ACME 9000 RI detector. The columns were calibrated against seven poly(methyl methacrylate) (PMMA) standard samples (Polymer Lab, PMMA Calibration Kit, M-M-10). SEM images of the polymers were recorded by the scanning electron microscope of model: HRSEM SUPRA 40, ZEISS (Germany), to examine the morphology of the amphiphilic and star polymers (Fig. S1†). Samples for SEM analysis were prepared by making 1 mg mL−1 aqueous solutions of polymers, deposited 2 drops of the solutions on copper grids and subsequently dried them up under vacuum. Their root mean square roughness and average roughness were determined by AFM analysis. Sample (polymer) films were prepared by depositing 3 drops of 1 mg sample per 1 mL of water, on a neat glass slide (1 cm × 1 cm) and allowed for drying in vacuum oven at room temperature for 24 h before AFM observation. The AFM images were recorded in semi contact mode by using noncontact silicon cantilever (NSG10-DLC) and the imaging was done on NT-MDT Model Solver NEXT (Fig. S1†). Image processing was done by using NOVA Px 3.1.0 Rev 3880 software.
Self assembly study
Self assembly of alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7 to form micelles was observed by the 1H NMR spectral analysis done in deuterium oxide and the 1H NMR spectra were recorded on a JEOL JNM-ECZ500R/S1 FTNMR (500 MHz) at room temperature and reported in parts per million (δ) from residual solvent peak. Fluorescence measurements were carried out on Varian Cary Eclipse fluorescence spectrometer. A series of aqueous solutions of alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7 having concentrations ranging from 1 × 10−4 to 1 mg mL−1 were prepared by diluting the stock solutions of polymers in deionized water. A stock solution of pyrene in acetone was added to a series of vials and acetone was evaporated under nitrogen. The polymer solutions prepared were then added to the vials to get a final concentration of pyrene equal to 6 × 10−7 M in each vial. These samples were equilibrated overnight at room temperature and then excitation spectra of the solutions were recorded at an emission wavelength of 394 nm using a slit width of 5 nm (Fig. S2†). The ratio of the pyrene peak intensities of the excitation spectra at 337.07 nm (I3) and 333.07 nm (I1) was analyzed as a function of polymer concentration. The critical micelle concentration (cmc) value was calculated from the point of interception of the two tangent straight lines at low concentration. Hydrodynamic diameter measurements of the micelles formed by both amphiphilic and star polymers were carried out by using a DLS instrument (Zetasizer Nano ZS, Malvern Instrument Ltd., UK) equipped with a He–Ne laser beam at 658 nm. Minimum three measurements at 25 °C were made for each sample with an equilibrium time of 2 minutes before starting the measurement. Samples were prepared by mixing 50 μL of the polymer solution (1 mg mL−1 aqueous solution) diluted with 1.95 mL CH3OH followed by filtering through a 0.45 μm polytetrafluorethylene (PTFE) filter. TEM analysis of amphiphilic and star polymers was done by using TECNAI 20 G2-electron microscope operated at accelerating voltage 200 kV. The TEM samples were prepared by dipping the carbon coated copper grid into the aqueous solutions of polymers (1 mg mL−1) followed by the removal of extra solution with a filter paper.
Strategy followed for the synthesis of dendrimer like star polymer based on β-cyclodextrin, with ABC type miktoarm star polymeric arms is depicted in Schemes 1–3. This polymer was synthesized by the combination of controlled/living radical polymerization methods (ATRP, ROP and RAFT polymerization) and modular approach (click reaction).
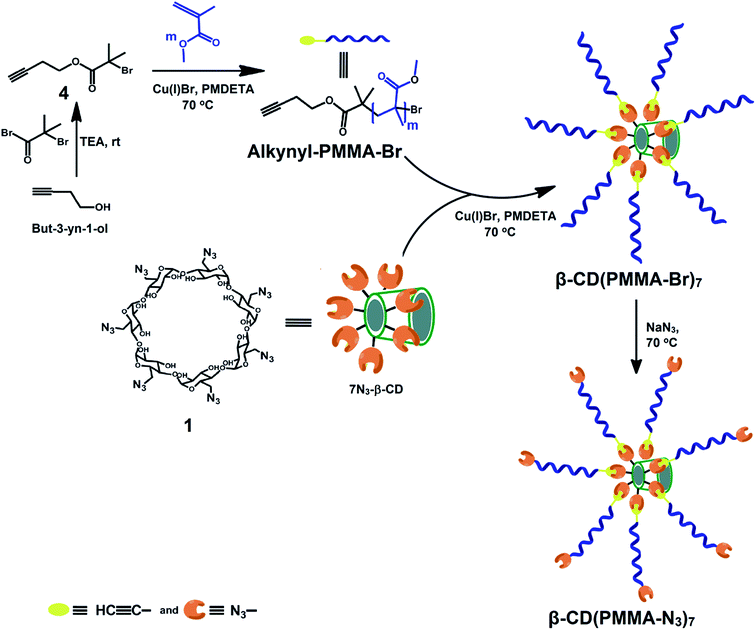 |
| | Scheme 1 Synthetic route of precursor star polymer (β-CD(PMMA-N3)7) and representation of its idealized structure. | |
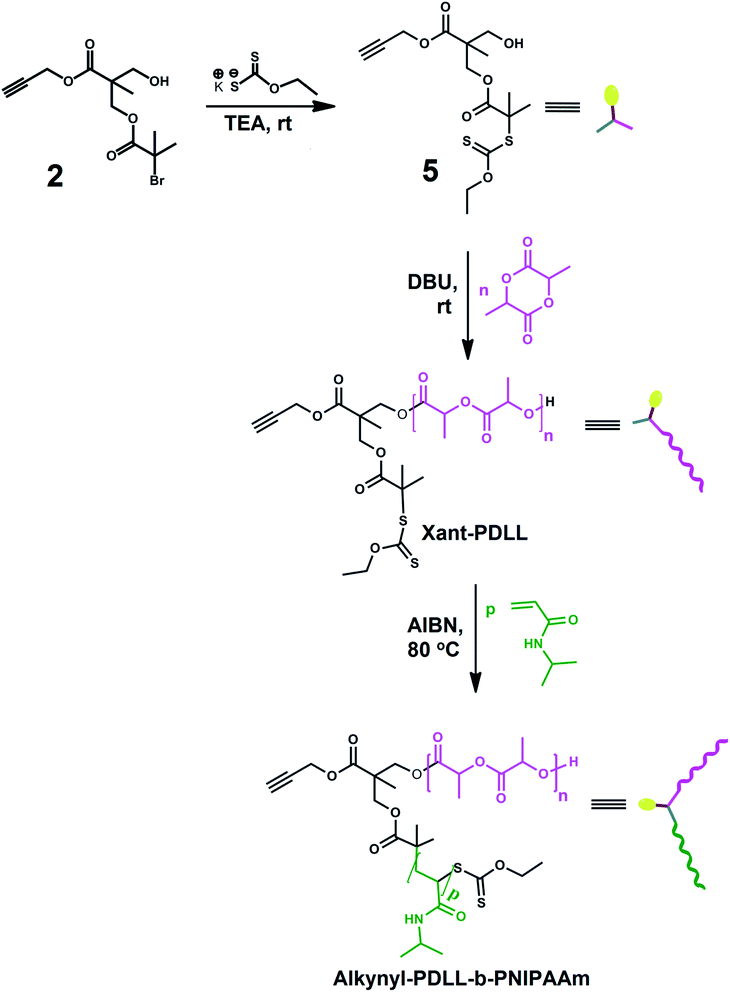 |
| | Scheme 2 Synthetic route of amphiphilic block copolymer i.e., alkynyl-PDLL-b-PNIPAAm. | |
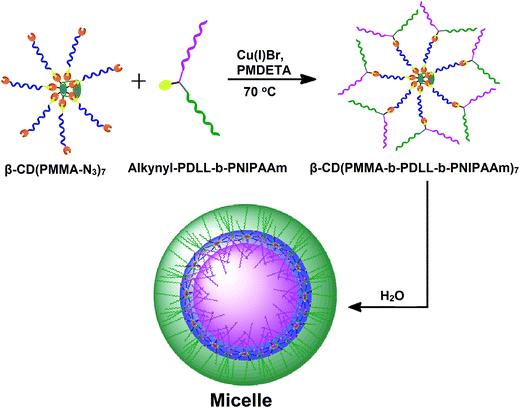 |
| | Scheme 3 Representation of idealized structures of β-CD(PMMA-b-PDLL-b-PNIPAAm)7, synthesized through the click reaction between β-CD(PMMA-N3)7 and alkynyl-PDLL-b-PNIPAAm. | |
Synthesis of but-3-yn-1-yl 2-bromo-2-methylpropanoate (alkynyl-Br) (4)
In a dried 250 mL round bottom flask, but-3-yn-1-ol (1.51 mL, 20 mmol) and triethyl amine (2.93 mL, 21 mmol) were dissolved in 15 mL of dichloro methane. The mixture was cooled to 0 °C on an ice bath and then 2-bromo-2-methylpropanoyl bromide (2.48 mL, 20 mmol) dissolved in 10 mL was added into the reaction mixture for 30 min which was followed by stirring for 16 h. Then the mixture was filtered and the filtrate was diluted with 50 mL of DCM. The solution was washed consecutively with saturated NaHCO3 solution (3 × 30 mL) and water (3 × 50 mL). The organic layer was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under vacuum and the product was purified by column chromatography with 1:10 ethyl acetate and hexane mixture. Yield: 86%. 1H NMR (300 MHz, CDCl3) (Fig. 1(a)): δ (ppm) = 4.2 (t, 2Hc), 2.6 (t, 2Hb), 2.0 (s, 1Ha + 6Hd).
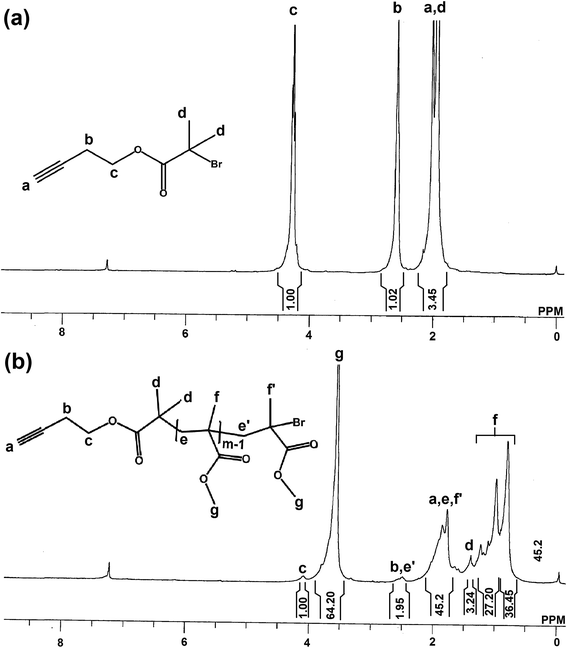 |
| | Fig. 1 1H NMR spectra of (a) alkynyl-Br and (b) alkynyl-PMMA-Br recorded in d-chloroform at room temperature. | |
Synthesis of alkyne terminated poly(methyl methacrylate) (alkynyl-PMMA-Br) through ATRP
Alkynyl-PMMA-Br was synthesized through ATRP of methyl methacrylate. MMA (5.3 mL, 50 mmol), CuBr (0.145 g, 1 mmol), but-3-yn-1-yl 2-bromo-2-methylpropanoate (0.219 g, 1 mmol) and PMDETA (0.210 mL, 1 mmol) were added to 3 mL of toluene in a 100 mL of Schlenk tube and the reaction mixture was degassed by three freeze–pump–thaw (FPT) cycles and left in vacuum. The Schlenk tube was subsequently placed in a thermostated oil bath at 70 °C until the reaction mixture became viscous. Then the darkgreen polymer mixture was diluted with THF, passed through a basic alumina column to remove the catalyst and precipitated in methanol. The polymer was dried for 24 h in a vacuum oven at 40 °C. Yield: 74%. [M]0/[I]0 = 50, [I]0:[CuBr]0:[PMDETA]0 = 1:1:1; (Mn,NMR = 4504 Da; Mn,GPC = 5500 Da; Mw/Mn = 1.51). 1H NMR (300 MHz, CDCl3) (Fig. 1(b)): δ (ppm) = 4.1 (br, 2Hc), 3.6 (br, 3mHg), 2.5 (br, 2Hb + 2He′), 1.9–1.8 (br, (2m-2)He + Ha + 3Hf′), 1.4 (s, 6Hd), 1.3–0.8 (br, (3m-3)Hf).
Synthesis of 7 arm star polymer precursor of poly(methyl methacrylate), based on β-CD (β-CD(PMMA-Br)7)
Click strategy, between 7N3-β-CD and alkynyl-PMMA-Br, was used to achieve the 7 arm star polymer of poly(methyl methacrylate), based on β-CD (β-CD(PMMA-Br)7) having bromine terminal on each arm. 7N3-β-CD (0.131 g, 0.1 mmol), Alkyne-PMMA-Br (3.468 g, 0.77 mmol) (calculated by using Mn,NMR = 4504 Da), CuBr (0.102 g, 0.7 mmol) and PMDETA (0.147 mL, 0.7 mmol) were dissolved in 10 mL of DMF in a 100 mL of Schlenk tube and the reaction mixture was degassed by three FPT cycles and left in vacuum. The tube was subsequently placed in a thermostated oil bath at 70 °C for 48 h. Then the reaction mixture was concentrated under vacuum and then dissolved in THF. The resulting solution was passed through basic alumina, and then concentrated under vacuum and precipitated in methanol. The precipitated polymer was dissolved in THF and gone through dialysis for 48 h to remove unclicked PMMA. Yield: 64%. (Mn,NMR = 19775 Da; Mn,GPC = 18000 Da; Mw/Mn = 2.08). 1H NMR (300 MHz, DMSO) (Fig. 2(a)): δ (ppm) = 8.4 (s, 4.1Ha), 5.6 (br, 7Hb′′ + 7He′′), 5.1 (br, 7Ha′′), 4.1–3.2 (br, 8.2Hc + 12.3mHg + 7Hc′′ + 7Hd′′ + 7Hf′′ + 7Hg′′ + 14Hh′′), 2.5 (br, 8.2Hb + 8.2He′), 1.8 (br, 4.1(2m-2)He + 12.3Hf′), 1.4 (br, 25.2Hd) and 1.2–0.8 (br, 12.3mHf).
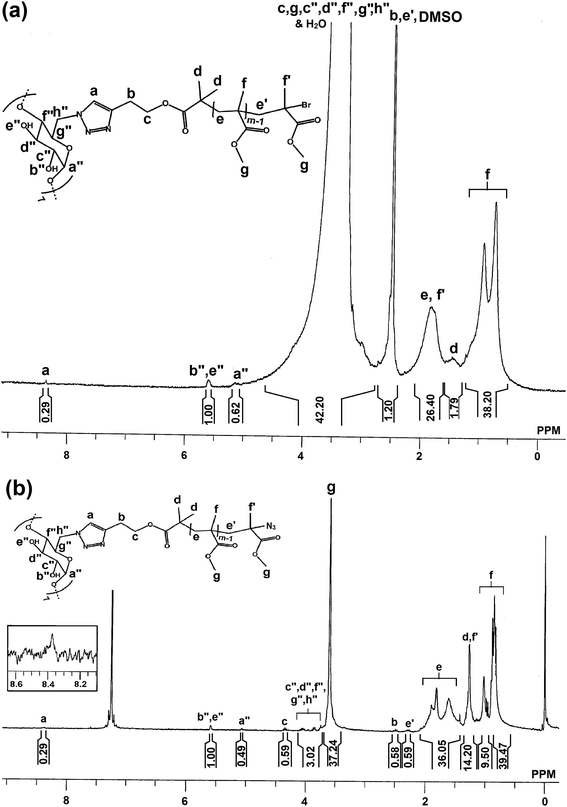 |
| | Fig. 2 1H NMR spectra of (a) β-CD(PMMA-Br)7 and (b) β-CD(PMMA-N3)7 done in d6-DMSO and d-chloroform, respectively at room temperature. | |
Synthesis of azide terminated 7 arm star polymer of poly(methyl methacrylate), based on β-CD (β-CD(PMMA-N3)7)
β-CD(PMMA-Br)7 (2.966 g, 0.15 mmol) (calculated by using Mn,NMR = 19775), and NaN3 (195 mg, 3 mmol) were dissolved in 10 mL of DMF and the reaction mixture was stirred in a thermostated oil bath at 70 °C for three days. The resulting mixture was concentrated under vacuum and precipitated in methanol. Yield: 91%. (Mn,NMR = 19619 Da; Mn,GPC = 18400 Da; Mw/Mn = 1.47). 1H NMR (300 MHz, CDCl3) (Fig. 2(b)): δ (ppm) = 8.4 (s, 4.1Ha), 5.6 (br, 7Hb′′ + 7He′′), 5.1 (br, 7Ha′′), 4.3 (t, 7.6Hc), 4.1–3.8 (m, 7Hc′′ + 7Hd′′ + 7Hf′′ + 7Hg′′ + 14Hh′′), 3.6 (br, 12.3mHg), 2.5 (t, 8.2Hb), 2.3 (br, 8.2He′), 1.8–1.5 (br, 4.1(2m-2)He), 1.2 (br, 24.6Hd + 12.3Hf′) and 1.0–0.8 (br, 12.3mHf).
Synthesis of mikto functional initiator (5)
A novel mikto functional initiator i.e., prop-2-yn-1-yl 3-((2-((ethoxycarbonothioyl)thio)-2-methylpropanoyl)oxy)-2-(hydroxymethyl)-2-methylpropanoate (5) was synthesized as follows: propargyl-3-[(2-bromo-2-methylpropanoyl)oxy]-2-(hydroxymethyl)-2-methylpropanoate (2) (2.385 g, 7.5 mmol) and triethyl amine (4.2 mL, 30 mmol) were dissolved in 40 mL of DCM and then potassium O-ethyl carbonodithioate (3) (2.775 g, 22.5 mmol) was added to the reaction mixture. After 16 h stirring at room temperature the mixture was filtered off and the filtrate was diluted with 25 mL of DCM. The solution was washed consecutively with saturated NH4Cl solution (3 × 50 mL), saturated NaHCO3 solution (3 × 50 mL) and water (3 × 100 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum and the product was purified by column chromatography with 1:100 ethyl acetate and hexane mixture. Yield: 76%. 1H NMR (300 MHz, CDCl3) (Fig. 3(a)): δ (ppm) = 4.8–4.5 (br, 2Hg + 2Hb + 2He), 4.4 (t, 1Hc), 4.0 (t, 1Hc′), 3.7 (br, 1Hi), 2.5 (s, 1Ha), 1.6–1.3 (br, 3Hd + 6Hf + 3Hh).
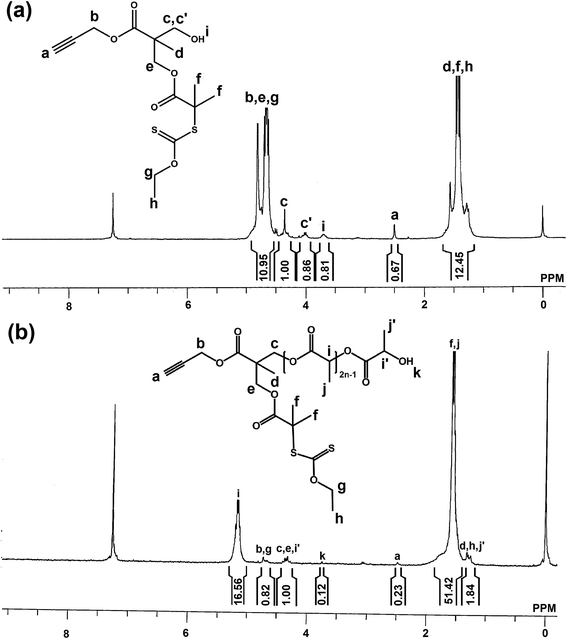 |
| | Fig. 3 1H NMR spectra of (a) mikto functional initiator, 5 and (b) Xant-PDLL in d-chloroform at room temperature. | |
ROP of D,L-lactide (Xant-PDLL) at room temperature
Above synthesized mikto functional initiator was employed in ring opening polymerization of D,L-lactide to synthesize poly(D,L-lactide) having xanthate and alkyne groups attached (Xant-PDLL). For this, mikto functional initiator (359 g, 1 mmol) and D,L-lactide (7.207 g, 50 mmol) were dissolved in 8 mL of DCM under nitrogen gas purging in a 100 mL of Schlenk tube. Then DBU (0.15 mL, 1 mmol) was added into the reaction mixture at room temperature. Within 5 minutes reaction mixture became viscous and then benzoic acid (134 mg, 1.1 mmol) was added to quench the polymerization. Then the reaction mixture was cooled in liquid nitrogen to stop polymerization, and dissolved in 50 mL of DCM and then washed with saturated NaHCO3 solution (3 × 50 mL) and water (3 × 100 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The resulting mixture was then precipitated in cold methanol to get poly(D,L-lactide) (Xant-PDLL). Yield: 78%. (Mn,NMR = 6408 Da; Mn,GPC = 7200 Da; Mw/Mn = 1.21). 1H NMR (300 MHz, CDCl3) (Fig. 3(b)): δ (ppm) = 5.2–5.1 (br, (2n-1)Hi), 4.7 (m, 2Hb + 2Hg), 4.4–4.3 (m, 2Hc + 2He + 1Hi′), 3.7 (s, 1Hk), 2.5 (s, 1Ha), 1.6–1.5 (br, (6n-3)Hj + 6Hf), 1.2 (m, 3Hd + 3Hh + 3Hj′).
RAFT polymerization of NIPAAm to synthesize alkyne terminated block copolymer of PDLL and PNIPAAm (alkynyl-PDLL-b-PNIPAAm)
Above synthesized poly(D,L-lactide) having xanthate and alkyne groups attached (Xant-PDLL) was used in synthesis of block copolymer of PDLL and PNIPAAm (alkynyl-PDLL-b-PNIPAAm). For this, in a 100 mL of Schlenk tube Xant-PDLL (3.849 g, 0.6 mmol) (calculated by using Mn,NMR = 6408) and NIPAAm (5.662 g, 50 mmol) were dissolved in 15 mL of THF under nitrogen purging. Then AIBN (0.025 g, 0.15 mmol) was added to the reaction mixture and the Schlenk tube was placed in a thermostated oil bath at 80 °C for 24 h. The reaction mixture became viscous and was concentrated under vacuum. The product was dissolved in 50 mL of DCM and washed with saturated NH4Cl solution (3 × 50 mL), saturated NaHCO3 solution (3 × 50 mL) and water (3 × 100 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The resulting mixture was then precipitated in hexane to get alkyne terminated block copolymer of PDLL and PNIPAAm (alkynyl-PDLL-b-PNIPAAm). Yield: 85%. (Mn,NMR = 11504 Da; Mn,GPC = 11600 Da; Mw/Mn = 1.19). 1H NMR (300 MHz, CDCl3) (Fig. 4): δ (ppm) = 6.5 (br, pHk), 5.2 (br, 2nHd), 4.8 (br, 2Hb + 2Hv), 4.3 (m, 2Hc + 2Hg + Hd′), 4.0 (br, pHt), 2.5 (s, 1Ha), 2.1 (br, pHj + 2Hi′), 1.8–1.3 (br, 2pHi + 6nHe + 6Hh), 1.2 (br, 6pHu + 3Hf + 3Hw + 3He′).
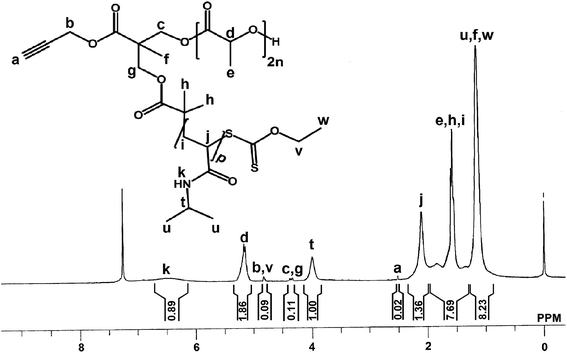 |
| | Fig. 4 1H NMR spectra of amphiphilic block copolymer, alkynyl-PDLL-b-PNIPAAm in d-chloroform at room temperature. | |
Synthesis of star polymer based on β-CD, having ABC type miktoarms (β-CD(PMMA-b-PDLL-b-PNIPAAm)7)
Star polymer based on β-cyclodextrin, in which the primary alcoholic arms of β-cyclodextrin linked to ABC type miktoarm star polymers was synthesized by the click reaction of β-CD(PMMA-N3)7 with alkynyl-PDLL-b-PNIPAAm.
For this, β-CD(PMMA-N3)7 (0.393 g, 0.02 mmol) (calculated by using Mn,NMR = 19619), alkynyl-PDLL-b-PNIPAAm (1.841 g, 0.16 mmol) (calculated by using Mn,NMR = 11504), CuBr (0.015 g, 0.1 mmol) and PMDETA (0.021 mL, 0.1 mmol) were dissolved in 5 mL of DMF in a 50 mL of Schlenk tube and the reaction mixture was degassed by three FPT cycles and left under vacuum. The tube was subsequently placed in a thermostated oil bath at 70 °C for 72 h. Then the reaction mixture was concentrated under vacuum and then dissolved in THF. The resulting solution was passed through basic alumina, and concentrated under vacuum. The polymer mixture obtained was subjected to dialysis for 48 h in a membrane of 12000–14000 Da cut off. Then the star polymer was precipitated in hexane. Yield: 71%. (Mn,NMR = 57352 Da; Mn,GPC = 69600 Da; Mw/Mn = 1.37). 1H NMR (300 MHz, CDCl3) (Fig. 5): δ (ppm) = 8.4 (s, 4.1Hi′), 7.6 (s, 3.28Ha), 6.5 (br, 3.28pHk), 5.9 (br, 7Hc′ + 7He′), 5.2 (br, 3.28(2n-1)Hd + 7Ha′), 4.7 (br, 6.56Hb + 6.56Hv), 4.3 (m, 6.56Hc + 6.56Hg + 8.2Hk′ + 3.28Hd′′), 4.0 (br, 7Hb′ + 7Hd′ + 7Hf′ + 7Hg′ + 14Hh′ + 3.28pHt), 3.6 (br, 12.3mHw′ + 3.28Hj′′), 2.4–2.1 (br, 3.28(p-1)Hj + 8.2Hj′ + 8.2 Hu′′ + 6.56Hi′′), 1.7–1.4 (br, 4.1(m-1)Hu′ + 6.56pHi + 9.84(2n-1)He + 19.68Hh), 1.2 (br, 19.68pHu + 9.84Hf + 9.84Hw + 24.6Ht′), 0.8 (br, 12.3mHv′ + 9.84He′′).
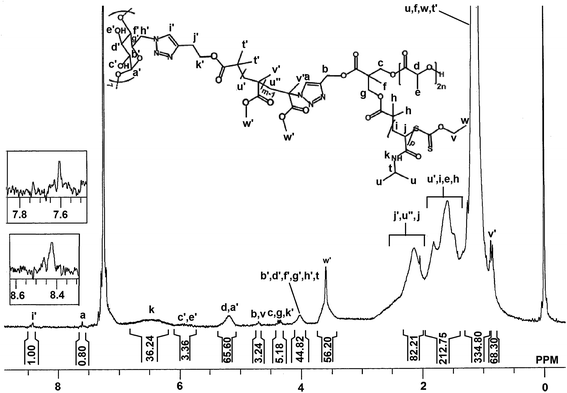 |
| | Fig. 5 1H NMR spectra of star polymer, β-CD(PMMA-b-PDLL-b-PNIPAAm)7 in d-chloroform at room temperature. | |
Results and discussion
The star polymer based on β-cyclodextrin having ABC type miktoarm star polymeric arms was synthesized by the combination ATRP, ROP and RAFT polymerization, and click reaction.
Synthesis of azide terminated star polymer based on β-CD and PMMA (β-CD(PMMA-N3)7)
But-3-yn-1-yl 2-bromo-2-methylpropanoate (alkynyl-Br) was synthesized by reacting but-3-yn-1-ol with 2-bromo-2-methylpropanoyl bromide in presence of TEA as base and DCM as solvent at room temperature. Thus synthesized alkynyl-Br was purified by using column chromatography and good yield was observed. In the 1H NMR of alkynyl-Br (Fig. 1(a)), done by using CDCl3 as solvent, a peak at 2.0 ppm due to the presence of both HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– (a) and six –CO(CH3)2Br (d) protons confirmed its formation.39 Thus synthesized, alkynyl-Br was used as initiator in ATRP of MMA to produce alkynyl-PMMA-Br. In the 1H NMR spectra (Fig. 1(b)), six methyl (d) protons of initiator were shifted from 2 ppm to 1.4 ppm during ATRP of MMA. Signals observed at 3.6, 1.9–1.8 and 1.3–0.8 ppm attributed to –COOCH3 (g), –CH2–CH (e) and –CH–CH3 (f) protons of PMMA respectively, confirmed the polymerization of MMA. The number of repeating units was calculated by comparing the peak area of methyl (g) protons of ester links present in PMMA, observed at 3.6 ppm, with that of methylene (c) protons of initiator at 4.1 ppm, and was found to be 42.8. The number average molecular weight estimated through 1H NMR (Mn,NMR) was 4504 Da. Mn,GPC and polydispersity index (PDI), were 5500 Da and 1.51 respectively.
C– (a) and six –CO(CH3)2Br (d) protons confirmed its formation.39 Thus synthesized, alkynyl-Br was used as initiator in ATRP of MMA to produce alkynyl-PMMA-Br. In the 1H NMR spectra (Fig. 1(b)), six methyl (d) protons of initiator were shifted from 2 ppm to 1.4 ppm during ATRP of MMA. Signals observed at 3.6, 1.9–1.8 and 1.3–0.8 ppm attributed to –COOCH3 (g), –CH2–CH (e) and –CH–CH3 (f) protons of PMMA respectively, confirmed the polymerization of MMA. The number of repeating units was calculated by comparing the peak area of methyl (g) protons of ester links present in PMMA, observed at 3.6 ppm, with that of methylene (c) protons of initiator at 4.1 ppm, and was found to be 42.8. The number average molecular weight estimated through 1H NMR (Mn,NMR) was 4504 Da. Mn,GPC and polydispersity index (PDI), were 5500 Da and 1.51 respectively.
Alkynyl-PMMA-Br, thus synthesized, was employed in click reaction with 7N3-β-CD to synthesize 7 arm star polymer of PMMA, based on β-CD, with bromide end groups i.e., β-CD(PMMA-Br)7. In the 1H NMR spectrum of β-CD(PMMA-Br)7 (Fig. 2(a)), done in DMSO, a signal observed at 8.3 ppm was due to the aromatic triazole ring proton, a, of PMMA part shifted towards low shield region, after the click reaction. The number of PMMA units linked to 7N3-β-CD through click reaction was calculated by comparing the peak area of secondary hydroxyl protons, b′′ and e′′, observed at 5.6 ppm with that of HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– proton of triazole ring observed at 8.3 ppm, and was found as 4.1. Calculated Mn,NMR, Mn,GPC and PDI were 19775 Da, 18000 Da and 2.08 respectively. Thus obtained, β-CD(PMMA-Br)7 was treated with sodium azide to synthesize β-CD(PMMA-N3)7 having azide end groups. The 1H NMR spectrum of β-CD(PMMA-N3)7 (Fig. 2(b)), done in CDCl3 revealed the appearance of a new signal at 1.2 ppm and smoothening of peak at 1.9 ppm, due to the shifting of methyl protons (f′), adjacent to azide groups, of terminal repeating unit of PMMA.
C– proton of triazole ring observed at 8.3 ppm, and was found as 4.1. Calculated Mn,NMR, Mn,GPC and PDI were 19775 Da, 18000 Da and 2.08 respectively. Thus obtained, β-CD(PMMA-Br)7 was treated with sodium azide to synthesize β-CD(PMMA-N3)7 having azide end groups. The 1H NMR spectrum of β-CD(PMMA-N3)7 (Fig. 2(b)), done in CDCl3 revealed the appearance of a new signal at 1.2 ppm and smoothening of peak at 1.9 ppm, due to the shifting of methyl protons (f′), adjacent to azide groups, of terminal repeating unit of PMMA.
Synthesis of amphiphilic block copolymer of PDLL and PNIPAAm having alkyne terminal (alkynyl-PDLL-b-PNIPAAm)
Mikto functional initiator (5) was synthesized by the reaction of propargyl-3-[(2-bromo-2-methylpropanoyl)oxy]-2-(hydroxymethyl)-2-methylpropanoate with potassium O-ethyl carbonodithioate in presence of TEA, as a base. The 1H NMR spectral analysis (Fig. 3(a)) revealed that, during the synthesis of compound 5 from 1, the six protons of –C(O)–C(CH3)2–S (“f” protons) shifted from 1.9 ppm to 1.4 ppm, and new signals of –C(S)–O–CH2– (g) and –O–CH2–CH3 (h) appeared at 4.8 and 1.3 ppm respectively. Thus 1H NMR confirmed the synthesis of mikto functional initiator, 5. Initiator, thus synthesized, was used in ring opening polymerization of D,L-lactide, through its hydroxyl end group, to obtain Xant-PDLL. Synthesis of Xant-PDLL was confirmed by its 1H NMR spectrum (Fig. 3(b)) containing the peaks of –CH (i) and –CH3 (j) proton signals of poly(D,L-lactide) appearing at 5.2–5.1 and 1.6–1.5 ppm respectively, along with initiator proton signals. The number of repeating units of DLL was estimated by comparing the peak area of CH, i, protons at 5.1 ppm with that of c, e and i′ protons at 4.3 ppm and was found as 41.9. Thus calculated Mn,NMR was 6408 Da, and Mn,GPC and PDI were 7200 Da and 1.21 respectively. Xant-PDLL, thus synthesized had alkyne and xanthate end groups for further processing.
Xant-PDLL was then employed in RAFT polymerization of NIPAAm through its xanthate end group, using AIBN as initiator to synthesize amphiphilic block copolymer (alkynyl-PDLL-b-PNIPAAm) with alkyne end group. Synthesis of alkynyl-PDLL-b-PNIPAAm, was confirmed by the 1H NMR spectral analysis done in CDCl3 (Fig. 4). Six protons of –OC(O)–C(CH3)2– (h protons) were shifted from 1.3 to 1.6 ppm after RAFT polymerization. New signals at 6.5, 4.0, 2.1, 1.8–1.3 and 1.2 ppm appeared due to the presence of repeating units of PNIPAAm. The number of repeating units of NIPAAm was calculated by comparing the peak area of –CH (t) protons, adjacent to –NH group, of NIPAAm units observed at 4.0 ppm with that of –CH (d) protons of PDLL part at 5.1 ppm, and was found as 45. Mn,NMR, thus estimated, was 11504 Da. Further, the GPC spectrum of alkynyl-PDLL-b-PNIPAAm contained a unimodal peak portraying the presence of amphiphilic block copolymer alone, with number average molecular weight (Mn,GPC) and PDI equals to 11600 Da and 1.19 respectively.
Synthesis of star polymer having ABC type polymeric miktoarms
The star polymer of interest i.e., β-CD(PMMA-b-PDLL-b-PNIPAAm)7 was finally attained through the click reaction between β-CD(PMMA-N3)7 and excess amounts of alkynyl-PDLL-b-PNIPAAm. Resulting polymeric mixture was run through basic alumina column to remove Cu(I)Br and its complexes. Unclicked amphiphilic block copolymer was then removed by using dialysis method. The aromatic proton of newly formed triazole ring, after the click reaction, appeared at 7.6 ppm (Fig. 5). Along with this the signals of all the protons of polymeric arms (PMMA, PDLL and PNIPAAm) and core (β-CD) were also present. The Mn,NMR estimated by comparing the peak area of preliminary triazole ring protons appeared at 8.4 ppm with the triazole ring protons appeared at 7.6 ppm was found to be 57352 Da. Thus 1H NMR spectral analysis, done in CDCl3, confirmed the synthesis of β-CD(PMMA-b-PDLL-b-PNIPAAm)7. GPC analysis (Mn,GPC = 69600 Da) also supported the synthesis of star polymer (Fig. 6). In the GPC analysis, the presence of traces was due to the unrevoked amounts of star polymer in which one unit of amphiphilic block copolymer is attached with the precursor star polymer. The equivalents of monomers used, number average molecular weights calculated from 1H NMR spectra and GPC techniques and polydispersity indices of the synthesized polymers are listed in Table 1.
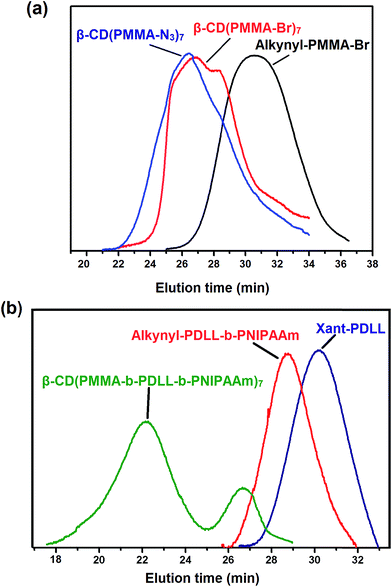 |
| | Fig. 6 (a) Gel permeation chromatograms of alkynyl-PMMA-Br, β-CD(PMMA-Br)7 and β-CD(PMMA-N3)7, and (b) Xant-PDLL, alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7. | |
Table 1 The synthetic and molecular weight details of the synthesized polymers
| Polymer |
Monomer (equivalents) |
Yield (%) |
Mn (NMR) (Da) |
Mn (GPC) (Da) |
PDI |
| The number of repeating units = 2 × (peak area of methyl (g) protons of ester links present in PMMA observed at 3.6 ppm)/3 × (methylene (c) protons of initiator at 4.1 ppm). The number of PMMA units linked to 7N3-β-CD through click reaction = 14 × (HCC– proton of triazole ring observed at 8.3 ppm)/(peak area of secondary hydroxyl protons (b′′, e′′) observed at 5.6 ppm). The number of repeating units of DLL = 5 × (peak area of CH (i) protons at 5.1 ppm)/2 × (peak area of c, e and i′ protons at 4.3 ppm). The number of repeating units of NIPAAm = 83.8 × (peak area of –CH (t) protons, adjacent to –NH group, of NIPAAm units observed at 4.0 ppm)/(peak area of –CH (d) protons of PDLL part at 5.1 ppm). The number of amphiphilic block copolymer units linked to β-CD(PMMA-N3)7 through click reaction = 4.1 × (peak area of triazole ring protons appeared at 7.6 ppm)/(peak area of preliminary triazole ring protons appeared at 8.4 ppm). |
| Alkynyl-PMMA-Br |
50 |
74 |
4504a |
5500 |
1.51 |
| β-CD(PMMA-Br)7 |
— |
64 |
19775b |
18000 |
2.08 |
| β-CD(PMMA-N3)7 |
— |
91 |
19619 |
18400 |
1.47 |
| Xant-PDLL |
50 |
78 |
6408c |
7200 |
1.21 |
| Alkynyl-PDLL-b-PNIPAAm |
50 |
85 |
11504d |
11600 |
1.19 |
| β-CD(PMMA-b-PDLL-b-PNIPAAm)7 |
— |
71 |
57352e |
69600 |
1.37 |
SEM and AFM analysis
The SEM images of alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7 are shown in ESI (Fig. S1(a) and (b)† respectively). The former one had granular surface where as the later one had smooth sheet like surface. Thus it was revealed from these images, that amphiphilic and star polymers have different surface morphologies. The respective root mean square roughness and average roughness, deduced from AFM analysis, of alkynyl-PDLL-b-PNIPAAm (Fig. S1(c)†) were 45.56 and 35.63 nm whereas of β-CD(PMMA-b-PDLL-b-PNIPAAm)7 (Fig. S1(d)†) were 30.85 and 21.46 nm. Thus the root mean square roughness and average roughness of alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7 were divergent from each other.
Self assembly study of the star polymers in aqueous solution
Nanostructured aggregates of the amphiphilic block copolymers can be formed by means of their self assembly in aqueous solutions. Due to the presence of both hydrophobic and hydrophilic parts, the synthesized star polymer could also form aggregations through self assembly. In case of amphiphilic block copolymer (alkynyl-PDLL-b-PNIPAAm) the 1H NMR spectral analysis done in deuterium oxide revealed the presence of PNIPAAm units only. In comparison with the 1H NMR spectrum attained in d-chloroform (Fig. 4), the peaks attributed to PDLL block totally disappeared in Fig. 7(a). It indicated that the self aggregates got formed in aqueous solution with PDLL blocks as the core and PNIPAAm blocks as the shell. Whereas, in case of star polymer (β-CD(PMMA-b-PDLL-b-PNIPAAm)7) for which the 1H NMR spectrum was attained in D2O (Fig. 7(b)), the peaks attributed to PDLL, PMMA and β-CD blocks i.e., hydrophobic parts totally disappeared when compared with Fig. 5. Only the signals attributed to PNIPAAm were present. This indicated that the nanostructured aggregates got formed in aqueous solution with PDLL, PMMA and β-CD blocks as the core and PNIPAAm blocks as the shell.
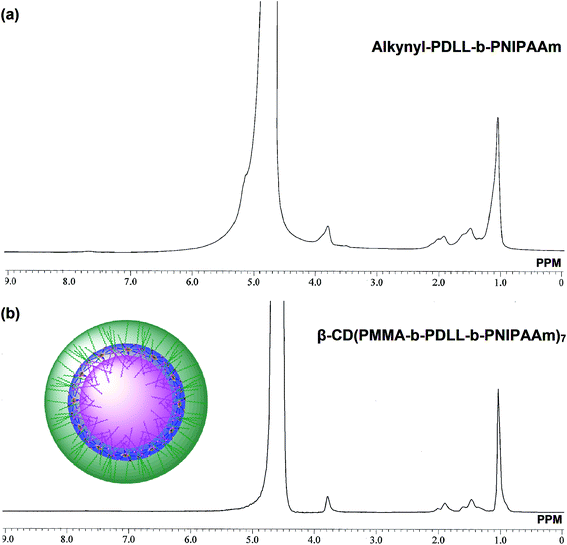 |
| | Fig. 7 1H NMR spectra of (a) amphiphilic block copolymer, alkynyl-PDLL-b-PNIPAAm and (b) star polymer, β-CD(PMMA-b-PDLL-b-PNIPAAm)7 in D2O at room temperature. | |
The plots between hydrodynamic diameter and intensity-weighted distribution, obtained from DLS measurement, of alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7 are shown in Fig. 8. The observed unimodal distribution functions indicated the formation of relatively similar size micellar aggregates and the respective hydrodynamic radii of amphiphilic and star polymers were ∼82.1 and ∼198.0 nm, and the respective polydispersity index (PDI) values were 0.428 and 0.395. Thus, it was revealed that the size of the micelle formed was greater in case of β-CD(PMMA-b-PDLL-b-PNIPAAm)7 than alkynyl-PDLL-b-PNIPAAm. This observation indicated that the hydrophobic part present in case of former one was more than the later one.
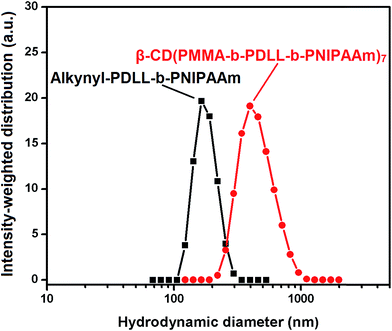 |
| | Fig. 8 Hydrodynamic diameter measurements alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7 carried out by using a DLS instrument equipped with a He–Ne laser beam at 658 nm. | |
The sizes of the micelles formed by alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7 were also examined by TEM analysis (Fig. 9(a) and (b) respectively). The average radii of the micelles formed by alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-b-PDLL-b-PNIPAAm)7 were found to be ∼9 and ∼62 nm respectively. The size of the micelles formed by star polymers observed in case of TEM analysis was smaller than that of DLS analysis. This might be because of the collapse of the micellar structure on TEM grids due to the dehydration.40 All these results confirmed the amphiphilic character of the synthesized star polymer.
 |
| | Fig. 9 TEM image of the micelles obtained from aqueous solution of (a) alkynyl-PDLL-b-PNIPAAm and (b) β-CD(PMMA-b-PDLL-b-PNIPAAm)7 (1 mg mL−1). | |
Conclusion
PMMA having alkyne group terminal, synthesized through ATRP, was clicked to 7N3-β-CD, yielding a star polymer of PMMA, based on β-CD. To this azidation was done in order to make the polymer clickable with alkynyl-PDLL-b-PNIPAAm. Parallelly, alkynyl-PDLL-b-PNIPAAm, an amphiphilic block copolymer of PDLL and PNIPAAm, was synthesized by the combination of ROP and RAFT polymerization techniques. Click reaction between alkynyl-PDLL-b-PNIPAAm and β-CD(PMMA-N3)7 yielded β-CD(PMMA-b-PDLL-b-PNIPAAm)7, a new star polymer whose 7 arms were linked to ABC type miktoarm star polymers. The 1H NMR spectra and GPC analysis confirmed the synthesis of all the initiators and polymers. The morphology deduced by SEM and roughness by AFM analysis supported the synthesis of amphiphilic and star polymers. These characteristics supported the synthesis of star polymer with polymeric miktoarms. 1H NMR spectral analysis done in D2O and fluorescence spectroscopic analysis of amphiphilic and star polymers revealed their self assembly property to form micelles. The cmc of the star polymer was observed to be greater than amphiphilic block copolymer. DLS and TEM analyses proved that the micellar size of star polymer is greater than that of amphiphilic block copolymer. The average radius of the micelles of star polymers deduced by TEM analysis was ∼62 nm.
Acknowledgements
The authors are grateful to UGC, New Delhi, India, for the financial support. The authors are grateful to Prof. Biswajit Ray for GPC analysis.
References
- Y. Satoh, K. Miyachi, H. Matsuno, T. Isono, K. Tajima, T. Kakuchi and T. Satoh, Macromolecules, 2016, 49, 499–509 CrossRef CAS.
- S. Ito, R. Goseki, T. Ishizone, S. Senda and A. Hirao, Macromolecules, 2013, 46, 819–827 CrossRef CAS.
- U. S. Gunay, H. Durmaz, E. Gungor, A. Dag, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 729–735 CrossRef CAS.
- N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 857–871 CrossRef CAS.
- K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171–1185 RSC.
- E. Blasco, B. V. K. J. Schmidt, C. B. Kowollik, M. Pinnol and L. Oriol, Macromolecules, 2014, 47, 3693–3700 CrossRef CAS.
- B. Li, L. Zhao, H. J. Qian and Z. Y. Lu, Soft Matter, 2014, 10, 2245–2252 RSC.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 11828–11834 CrossRef CAS PubMed.
- G. M. Soliman, A. Sharma, D. Maysinger and A. Kakkar, Chem. Commun., 2011, 47, 9572–9587 RSC.
- A. A. Steinschulte, B. Schulte, S. Rutten, T. Eckert, J. Okuda, M. Moller, S. Schneider, O. V. Borisovdef and F. A. Plamper, Phys. Chem. Chem. Phys., 2014, 16, 4917–4932 RSC.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3588–3598 CrossRef CAS.
- K. Khanna, S. Varshney and A. Kakkar, Macromolecules, 2010, 43, 5688–5698 CrossRef CAS.
- A. Hanisch, H. Schmalz and A. H. E. Muller, Macromolecules, 2012, 45, 8300–8309 CrossRef CAS.
- D. Konkolewicz, M. J. Monteiro and S. Perrier, Macromolecules, 2011, 44, 7067–7087 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2008, 41, 4250–4257 CrossRef CAS.
- T. He, D. Li, X. Sheng and B. Zhao, Macromolecules, 2004, 37, 3128–3135 CrossRef CAS.
- J. A. Kalow and T. M. Swager, ACS Macro Lett., 2015, 4, 1229–1233 CrossRef CAS PubMed.
- A. Dag, H. Durmaz, V. Kirmizi, G. Hizal and U. Tunca, Polym. Chem., 2010, 1, 621–623 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- M. A. Quadir, M. Martin and P. T. Hammond, Chem. Mater., 2014, 26, 461–476 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- M. R. Whittaker, C. N. Urbani and M. J. Monteiro, J. Am. Chem. Soc., 2006, 128, 11360–11361 CrossRef CAS PubMed.
- G. M. Soliman, A. Sharma, Y. Cui, R. Sharma, A. Kakkar and D. Maysinger, J. Nanomed. Biother. Discovery, 2014, 4, 1–10 Search PubMed.
- K. V. Butsele, C. A. Fustin, J. F. Gohy, R. Jerome and C. Jerome, Langmuir, 2009, 25, 107–111 CrossRef PubMed.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1218–1228 CrossRef CAS.
- C. Ye, G. Zhao, M. Zhang, J. Du and Y. Zhao, Macromolecules, 2012, 45, 7429–7439 CrossRef CAS.
- H. Liu, W. Pan, M. Tong and Y. Zhao, Polym. Chem., 2016, 7, 1603–1611 RSC.
- E. Gungor, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4459–4468 CrossRef CAS.
- O. Altintas, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5916–5928 CrossRef CAS.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, Biomacromolecules, 2010, 11, 934–943 CrossRef CAS PubMed.
- U. Ali, K. J. B. A. Karim and N. A. Buang, Polym. Rev., 2015, 55, 678–705 CrossRef CAS.
- T. Isono, Y. Kondo, I. Otsuka, Y. Nishiyama, R. Borsali, T. Kakuchi and T. Satoh, Macromolecules, 2013, 46, 8509–8518 CrossRef CAS.
- H. Durmaz, A. Dag, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1195–1200 CrossRef CAS.
- C. S. Biswas, V. K. Patel, N. K. Vishwakarma, V. K. Tiwari, B. Maiti, P. Maiti, M. Kamigaito, Y. Okamoto and B. Ray, Macromolecules, 2011, 44, 5822–5824 CrossRef CAS.
- X. Huan, D. Wang, R. Dong, C. Tu, B. Zhu, D. Yan and X. Zhu, Macromolecules, 2012, 45, 5941–5947 CrossRef CAS.
- K. Ramesh, A. K. Mishra, V. K. Patel, N. K. Vishwakarma, C. S. Biswas, T. K. Paira, T. K. Mandal, P. Maiti, N. Misra and B. Ray, Polymer, 2012, 53, 5743–5753 CrossRef CAS.
- D. Vizitiu, C. S. Walkinshaw, B. I. Gorin and G. R. J. Thatcher, J. Org. Chem., 1997, 62, 8760–8766 CrossRef CAS.
- N. Mourtzis, K. Eliadou, C. Aggelidou, V. Sophianopoulou, I. M. Mavridisa and K. Yannakopoulou, Org. Biomol. Chem., 2007, 5, 125–131 CAS.
- T. Yamada and A. Takasu, RSC Adv., 2015, 5, 41445–41456 RSC.
- A. K. Mishra, V. K. Patel, N. K. Vishwakarma, C. S. Biswas, M. Raula, A. Misra, T. K. Mandal and B. Ray, Macromolecules, 2011, 44, 2465–2473 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fluorescence spectroscopic analysis, SEM and AFM images of the synthesized amphiphilic and dendritic star polymers. See DOI: 10.1039/c6ra09660c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 











