
Coordination polymer-derived mesoporous Co3O4 hollow nanospheres for high-performance lithium-ions batteries†
Abstract
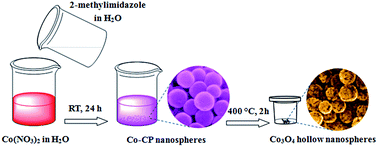
A green and convenient approach has been developed to fabricate mesoporous Co3O4 hollow nanospheres composed of nanosized building subunits, which involves a morphology-inherited and thermolysis-induced transformation of Co-based coordination polymers. When evaluated as anode materials for lithium-ion batteries, the as-fabricated hollow nanospheres exhibited excellent electrochemical performance with high reversible specific capacity, excellent cycling stability (851 mA h g−1 after 100 cycles) and exceptional rate capability (1053, 976, 887, and 695 mA h g−1 at the current densities of 0.3, 0.5, 1 and 5 A g−1, respectively).
 Please wait while we load your content...
Please wait while we load your content...

