
Retracted Article: Enhanced electrocatalytic activity and durability of highly monodisperse Pt@PPy–PANI nanocomposites as a novel catalyst for the electro-oxidation of methanol†
Abstract
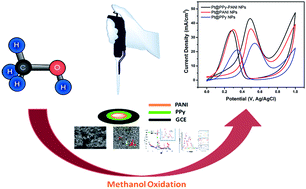
Highly monodisperse Pt nanocomposites (Pt@PPy–PANI NPs) supported on polypyrrole (PPy)–polyaniline (PANI) have been successfully synthesized for the first time by a simple one-pot process, which involves the simultaneous reduction of the conducting polypyrrole (PPy)–polyaniline (PANI) and Pt precursors using DMAB (dimethylamine borane) as a reductant under ultrasonic conditions. Pt@PPy–PANI NPs have been characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS). All the results show that highly crystalline and stable colloidal Pt@PPy–PANI NPs have been formed as one of the most active and long-lived catalysts with superior reusability performance for the electro-oxidation of methanol at room temperature. Compared to PPy or PANI supported Pt nanoparticles, Pt@PPy–PANI NPs exhibit extraordinary electrocatalytic activity and stability toward the electro-oxidation of methanol, showing their potential use as a new electrode material for direct methanol fuel cells (DMFCs). It should be primarily attributed to the PPy–PANI, which not only provided larger surface area and flaws for the deposition of Pt nanoparticles, but also the adsorption of more methanol molecules for further oxidation.
 Please wait while we load your content...
Please wait while we load your content...

