
Tailoring the electronic properties among oxoarsine, arsinoyl and arsine oxide isomers: the simplest molecular systems with an arsenic–oxygen bond†
Rommel B. Viana*
Departamento de Química e Física Molecular, Instituto de Química de São Carlos, Universidade de São Paulo, Av. Trabalhador São Carlense, 400 (caixa-postal: 780). Bairro: Centro, CEP 13560970, São Carlos, SP, Brazil. E-mail: rommelbv@yahoo.com.br
First published on 14th September 2016
Abstract
The main goal of this investigation is to understand the reaction pathways and the electronic and spectroscopy properties of AsOHn radicals (n = 0–3), which are some of the simplest compound models with an arsenic–oxygen bond. A CCSD(T) level of theory with a complete basis set limit (CBS) was applied to understand the relative stability and the reaction pathways among the isomers. Several reaction pathways such as the unimolecular rearrangement routes, the internal rotational transition state structures, and the hydrogen release routes were also evaluated among these structures. In the case of oxoarsine isomers, it was seen that the oxoarsine (HAsO) ground state structure presents a singlet state and AsOH possesses a triplet ground one. trans-Hydroxyarsinyl (trans-HAsOH) is the global minimum structure with an energy gap around 20 kcal mol−1 when compared with arsinoyl (H2AsO), and this energy difference is increased two times when compared with AsOH2. Arsinous acid (H2AsOH) is more stable than arsine oxide (H3AsO) based on the relative energy difference, while it is predicted that there is a large energy gap when compared with HAs(H2O) stereoisomers. The heat of formation was also calculated for each isomer. In addition, in the characterization of arsenic–oxygen bond characters, several bond order indexes and different population methods were also applied to understand the influence of different methodologies, as well as the Quantum Theory of Atoms in Molecules (QTAIM) and the Natural Bond Orbital (NBO) method.
1. Introduction
In the last four decades there has been tremendous interest in phosphine oxides,1–17 phosphinoyl18–24 and oxophosphine radicals25–33 at an experimental and computational level, while the same was also seen for their sulfur-analogues.34–46 Nevertheless, there is currently only limited information in the literature regarding arsine oxide, arsinoyl and oxoarsine isomers.47–57 Although there are some experimental47,52,53,56 and computational48–51,54,55 data in the literature from these arsenic-bearing radicals, there is an absence of information concerning the relative stability of the isomers as well as concerning the isomerization and hydrogen release pathways, which could shed light on future experimental and computational studies.A special case is oxoarsine (HAsO), which represents the simplest compound model for an analysis in the As![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. The first detection of HAsO was report by Andrews et al.,47 which reported a tentative identification of oxoarsine among the photolysis products of arsine–ozone complexes in argon matrices. Twelve years later, Grimminger and Clouthier53 described the vibrational and rotational spectroscopic analysis of HAsO, which was combined with a quantum chemical investigation using the Coupled-Cluster theory. It is noteworthy to comment that HAsO presents a 1A′ ground state and it is a predicted AsOH isomer based on the results from analogous phosphorous-bearing radicals.27,28,31,34,40,46 Looking at the oxoarsine isomer (AsOH), it is important to mention that Monahan-Pendergast et al.50 reported an ab initio investigation where they demonstrated that AsOH is a potential radical that can be released from coal combustion into the atmosphere. Nevertheless, based in the results of Monahan-Pendergast et al.,50 the AsOH ground state presents a singlet state, while based on phosphorus-analogous structures [HPX/PXH (X = O, S)]27,28,31,34,40,46 it was seen that the HPX ground state structures present singlet states and the PXH isomers possess triplet ground states. Therefore, a question that arises from these results reported for the phosphorous analogous structures27,28,31,34,40,46 is whether AsOH presents a triplet ground state and what the magnitude of the energy gap between singlet and triplet states would be.
O bond. The first detection of HAsO was report by Andrews et al.,47 which reported a tentative identification of oxoarsine among the photolysis products of arsine–ozone complexes in argon matrices. Twelve years later, Grimminger and Clouthier53 described the vibrational and rotational spectroscopic analysis of HAsO, which was combined with a quantum chemical investigation using the Coupled-Cluster theory. It is noteworthy to comment that HAsO presents a 1A′ ground state and it is a predicted AsOH isomer based on the results from analogous phosphorous-bearing radicals.27,28,31,34,40,46 Looking at the oxoarsine isomer (AsOH), it is important to mention that Monahan-Pendergast et al.50 reported an ab initio investigation where they demonstrated that AsOH is a potential radical that can be released from coal combustion into the atmosphere. Nevertheless, based in the results of Monahan-Pendergast et al.,50 the AsOH ground state presents a singlet state, while based on phosphorus-analogous structures [HPX/PXH (X = O, S)]27,28,31,34,40,46 it was seen that the HPX ground state structures present singlet states and the PXH isomers possess triplet ground states. Therefore, a question that arises from these results reported for the phosphorous analogous structures27,28,31,34,40,46 is whether AsOH presents a triplet ground state and what the magnitude of the energy gap between singlet and triplet states would be.
He et al.52 provided the first spectroscopic characterization of the arsinoyl radical (H2AsO) in the gas phase using a combination of laser-induced fluorescence and single vibronic level emission spectroscopy in a supersonic expansion. In this context, Tarroni and Clouthier51 calculated the spectroscopic properties of the first two excited states of H2AsO employing different quantum chemical methodologies, while Stojanovic55 reported the H2AsO isotropic and anisotropic hyperfine parameters with the use of various Density Functional Theory (DFT), MP2 and CCSD methods. It is noteworthy to comment that H2AsO possesses three possible isomers [AsOH2 and hydroxyarsinyl stereoisomers (cis/trans)], and most of the information in the literature is centered around arsinoyl free radicals. Based on the results for the thiophosphine analogues, it can be seen that trans-thiohydroxyphosphinyl (trans-HPSH) is the most stable isomer.20,40
The first identification of arsine oxide (H3AsO) and its isomers [arsinous acid, H2AsOH (cis/trans)] in the gas phase were performed by Andrews et al.47 in 1989, which demonstrated that both radicals are products of the photolysis of the AsH3–O3 complex. Schneider et al.48 applied a Hartree–Fock (HF) calculation level to calculate several spectroscopic parameters of both isomers one year later, while Dobado et al.10 applied the Quantum Theory of Atoms in Molecules (QTAIM) to analyze the arsenic-oxygen bonding properties of these two structures. Sola and Toro-Labbe49 showed that the energy gap between trans-H2AsOH and H3AsO is 29.4 kcal mol−1, demonstrating that arsinous acid is the most stable, while the H3AsO → H2AsOH isomerization barrier height is 51.3 kcal mol−1 using the B3LYP/6-31+G(d)//HF/6-31G(d) method. In this aspect, it is important to comment that these investigations10,48,49 set aside the metastable arsenyl HAs(H2O) stereoisomers. Nevertheless, phosphorus analogous were seen to have a large energy gap between HP(H2X) and H3PX isomers.13,35,39,40 Furthermore, Orthaber et al.54 evaluated the substituent effect in the arsine oxide structure and also showed the reaction mechanism between H3AsO and H2S. In addition, Morris et al.56 observed the H2AsOH–CrCl2O complex in argon matrices produced by the thermal and photochemical reaction between CrCl2O2 and arsine.
The purpose of this study is to perform quantum chemical analysis of the arsenic oxide radicals. This study analyzed several AsOHn isomers (n = 1–3), including HAsO, AsOH, HAsOH (cis/trans), H2AsO, AsOH2, H3AsO, H2AsOH (cis/trans) and HAs(H2O) (cis/trans). Among the reaction pathways, different mechanisms were considered as the isomerization routes, the internal rotational transition states which connect the stereoisomers, as well as the hydrogen release routes. In addition, a topological investigation employing the Quantum Theory of Atoms in Molecules (QTAIM), the Natural Bond Orbital (NBO) method, different bond indexes and several population methodologies were performed in this study for the analysis of the different arsenic–oxygen bond characters. The heat of formation was also provided for each AsOHn isomer.
2. Methodology
Stationary points on the potential energy surface were characterized by an evaluation of their harmonic vibrational frequencies. The absence of imaginary frequencies indicated their nature as minima. Geometric optimization routines were performed with the quadratic configuration interaction with single and double excitations (QCISD)58,59 using the cc-pVTZ basis sets,60,61 employing a tight convergence for the optimization and for the self-consistent field (SCF). For some analysis, the B3LYP functional62–64 and the cc-pVTZ basis sets60,61 were also used, however in these cases a ultrafine grid was also applied in addition to the tight convergence in the optimization and for SCF. The transition state structures were confirmed by a harmonic frequency analysis and all connections were evaluated by the intrinsic reaction coordinate (IRC) calculations.65,66 Moreover, aiming to evaluate the multi-reference character of these systems, T1 (ref. 67) and Q1 (ref. 68) diagnostics were employed using the single excitations on the CCSD and QCISD method with cc-pVTZ basis sets, respectively. These calculations were performed on the geometries obtained with QCISD/cc-pVTZ. The results for both diagnostic methods varied from 0.009 to 0.043. At this point, it is noteworthy to mention that values lower than 0.045 by T1 diagnostic can be analyzed with single reference wave function methods,69 while a similar feature is also reported by other systems using the Q1 diagnostic.70,71In order to refine the electronic energy, initially the Hartree–Fock basis set limit energy was determined with the three-point extrapolation formula of Peterson et al.72 using the aug-cc-pVnZ (n = T, Q, 5) basis sets. The correlation energy was estimated at CCSD(T) level73 with the two-point extrapolation formula of Halkier et al.74 using the aug-cc-pVnZ (n = T, Q) basis sets. The core-correlation was carried out by the energy difference between all electrons and the frozen-core approximation at the CCSD(T) level using the cc-pVTZ basis sets. The scalar relativistic correction was determined using the Douglas–Kroll–Hess second-order scalar relativistic method75,76 and it was calculated by the difference between the CCSD(T)/cc-pVTZ-DK77 and CCSD(T)/cc-pVTZ results. The perturbative correction for quadruple excitations was obtained from the difference between the BD(TQ) and BD(T) methods78 using the cc-pVTZ basis sets. This multistep methodology will be referred hereafter as CCSD(T)/CBS.
Quantum Theory of Atoms in Molecules (QTAIM)79 and Natural Bond Orbital (NBO) analysis methods were used to provide an understanding of the arsenic–oxygen bond nature among the molecules studied here. QTAIM and NBO methodologies were carried out with the AIMALL80 and NBO6.0 (ref. 81) programs, respectively. QTAIM analysis was performed to consider the properties of the arsenic–oxygen bond critical points (BCPs) with coordinates (3, −1) among the arsenic bearing radicals as electronic charge density [ρ(r)] and its Laplacian [∇2ρ(r)], local potential energy [V(r)], local energy density [G(r)], total energy density [H(r)] and ellipticity (ε). In this respect, it is important to note that the Laplacian of the electronic energy with a negative value (∇2ρ < 0) is a characteristic of the concentration of charge density, and a positive value (∇2ρ > 0) indicates charge depletion. The relationship between the modulus of the local potential energy and the local energy density (|V(r)|/G(r)) can be used to determine the covalent nature of the interactions. |V(r)|/G(r) > 2 indicates a covalent interaction, and |V(r)|/G(r) < 1 indicates a non-covalent interaction. Another important parameter is the total energy density [H(r)]. H(r) is represented by the sum of V(r) and G(r). H(r) works as an index of the amount of covalency in the chemical interaction,82 where H(r) < 0 indicates an interaction with significant sharing of electrons, while H(r) > 0 is an indication of an interaction with an ionic nature.
The bond order index was carried out using three different indices as the bond delocalization index (BDI),83 the Wiberg bond index (bowi)84 and the Mayer bond order index (bomay).85 An analysis using Natural Resonance Theory (NRT)86–88 was also performed to assess the percentage weights among the main resonance structures of each isomer studied. NRT results were obtained by considering the mean values between α and β orbitals. To evaluate the atomic charge distribution, QTAIM and NPA89 charge method was employed. It is noteworthy to mention that QTAIM, NBO, NRT, bond indexes and population methodologies were calculated with the B3LYP/cc-pVTZ method.
In addition, G4 (ref. 90) and G4MP2 (ref. 91) methods were used to evaluate the heat of formation from each AsOHn radical. The methodology outlined by Curtiss et al.92 was used to predict the heat of formation, where the total atomization energy was made use of using the known experimental elemental heat of formation at 0 K:93 H (ΔH0 Kf = 171.29 kcal mol−1), O (ΔH0 Kf = 59.56 kcal mol−1). The heat of formation value recommended by Mok et al.94 for arsenic element was used (ΔH0 Kf = 70.30 kcal mol−1), while the enthalpy correction of 1.00 kcal mol−1 was attributed to this atom. The G4* and G4MP2* methods were also used, which is essentially the G4 (ref. 90) and G4MP2 (ref. 91) methods, however in these cases the ZPVE value of each method was replaced by the ZPVE obtained with QCISD/cc-pVTZ level without any scaling factor. A similar procedure was already seen in literature95 to increase the accuracy of composite methods in the estimation of experimental heat of formation. All of the electronic structure calculations were performed with the GAUSSIAN 09 quantum chemistry code.96
3. Results and discussions
The molecular structure of each AsOHn radical is shown in Fig. 1, along with the bond lengths and angles calculated with QCISD/cc-pVTZ. Several spectroscopic and electronic properties involving the arsenic–oxygen bond of each radical are demonstrated in Table 1. Table 2 reports several parameters obtained from topological analysis with QTAIM of arsenic–oxygen bond critical points (BCPs), and Fig. 2 presents contour maps of the Laplacian distribution of the electronic charge density [∇2ρ(r)]. Further, the heat of formation results calculated with G4 and G4MP2 methods are presented in Table 3.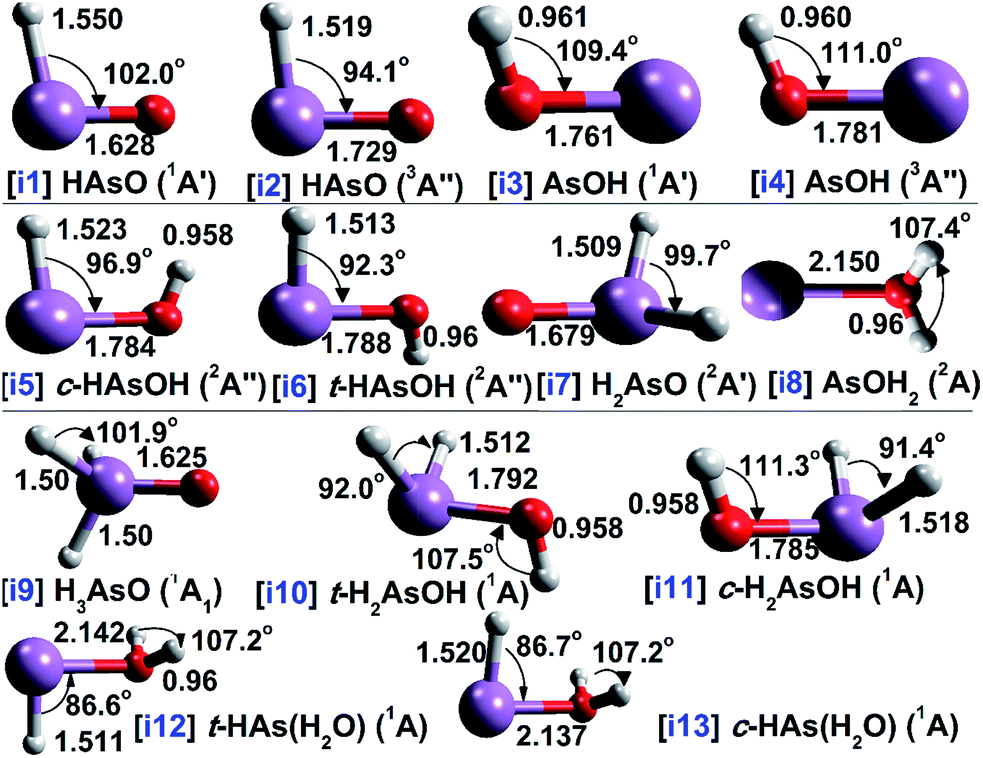 | ||
| Fig. 1 The oxoarsine (i1–i4), arsinoyl (i5–i8) and arsine oxide (i9–i13) isomer structures. The geometric parameters (bond lengths and angles in Å and °, respectively) were obtained with QCISD/cc-pVTZ. The pink ball refers to the arsenic atom, while the white and red balls refer to hydrogen and oxygen elements. | ||
| Radical | ν(As–O) | Atomic charge | Polarization | Bond order index | NRT | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| q(As) | q(O) | % As | % O | bowi | BDI | bomay | bonrt | % Cov | % Ion | ||
| AsO | 993.6 | 0.88 (0.95) | −0.88 (−0.95) | 22.2 | 77.8 | 1.59 | 1.76 | 2.03 | 1.39 | 44 | 56 |
| i1 | 988.3 | 1.07 (1.32) | −0.89 (−0.98) | 25.9 | 74.1 | 1.63 | 1.75 | 2.08 | 2.03 | 51 | 49 |
| i2 | 742.4 | 0.61 (0.90) | −0.57 (−0.70) | 20.9 | 79.1 | 1.03 | 1.09 | 1.26 | 1.82 | 42 | 58 |
| i3 | 742.4 | 0.44 (0.52) | −0.93 (−1.10) | 17.7 | 82.3 | 0.97 | 1.30 | 1.25 | 2.00 | 28 | 72 |
| i4 | 704.4 | 0.51 (0.56) | −0.99 (−1.14) | 15.4 | 84.6 | 0.74 | 1.02 | 1.80 | 1.52 | 30 | 70 |
| i5 | 699.6 | 0.62 (0.88) | −0.95 (−1.14) | 16.9 | 83.1 | 0.76 | 0.94 | 1.01 | 1.49 | 34 | 66 |
| i6 | 699.6 | 0.58 (0.86) | −0.94 (−1.13) | 17.2 | 82.8 | 0.77 | 0.97 | 1.02 | 1.50 | 34 | 66 |
| i7 | 867.1 | 0.89 (1.39) | −0.74 (−0.87) | 37.8 | 62.2 | 1.15 | 1.16 | 1.52 | 1.50 | 69 | 31 |
| i8 | 297.5 | −0.16 (−0.10) | −0.86 (−1.15) | 8.1 | 91.9 | 0.31 | 0.63 | 0.39 | 1.00 | 32 | 68 |
| i9 | 986.0 | 1.26 (1.94) | −1.02 (−1.11) | 32.5 | 67.5 | 1.21 | 1.31 | 1.86 | 1.13 | 57 | 43 |
| i10 | 685.9 | 0.66 (1.19) | −0.96 (−1.15) | 22.8 | 77.2 | 0.77 | 0.93 | 1.04 | 1.02 | 45 | 55 |
| i11 | 692.3 | 0.71 (1.23) | −0.97 (−1.16) | 22.3 | 77.7 | 0.76 | 0.90 | 1.02 | 1.01 | 44 | 56 |
| i12 | 310.1 | −0.09 (0.21) | −0.84 (−1.15) | 9.1 | 90.9 | 0.34 | 0.58 | 0.40 | 1.00 | 18 | 82 |
| i13 | 310.5 | −0.06 (0.23) | −0.84 (−1.15) | 9.0 | 91.0 | 0.34 | 0.94 | 0.39 | 1.00 | 18 | 82 |
| Radical | ρ(r) | ∇2ρ(r) | |V(r)|/G(r) | H(r) | ε |
|---|---|---|---|---|---|
| AsO | 0.2179 | 0.8939 | 1.41 | −0.1546 | 0.0214 |
| i1 | 0.2169 | 0.8222 | 1.43 | −0.1563 | 0.0651 |
| i2 | 0.1737 | 0.5161 | 1.47 | −0.1144 | 0.0906 |
| i3 | 0.1536 | 0.4659 | 1.44 | −0.0923 | 0.0236 |
| i4 | 0.1488 | 0.3921 | 1.48 | −0.0891 | 0.0781 |
| i5 | 0.1477 | 0.3649 | 1.49 | −0.0887 | 0.1194 |
| i6 | 0.1466 | 0.3579 | 1.50 | −0.0880 | 0.1082 |
| i7 | 0.1951 | 0.5803 | 1.49 | −0.1374 | 0.0048 |
| i8 | 0.0613 | 0.1671 | 1.24 | −0.0135 | 0.2570 |
| i9 | 0.2185 | 0.6322 | 1.51 | −0.1636 | 0.0000 |
| i10 | 0.1463 | 0.3128 | 1.53 | −0.0891 | 0.0414 |
| i11 | 0.1484 | 0.3295 | 1.52 | −0.0907 | 0.0392 |
| i12 | 0.0606 | 0.1596 | 1.25 | −0.0136 | 0.3052 |
| i13 | 0.1477 | 0.3649 | 1.49 | −0.0887 | 0.1194 |
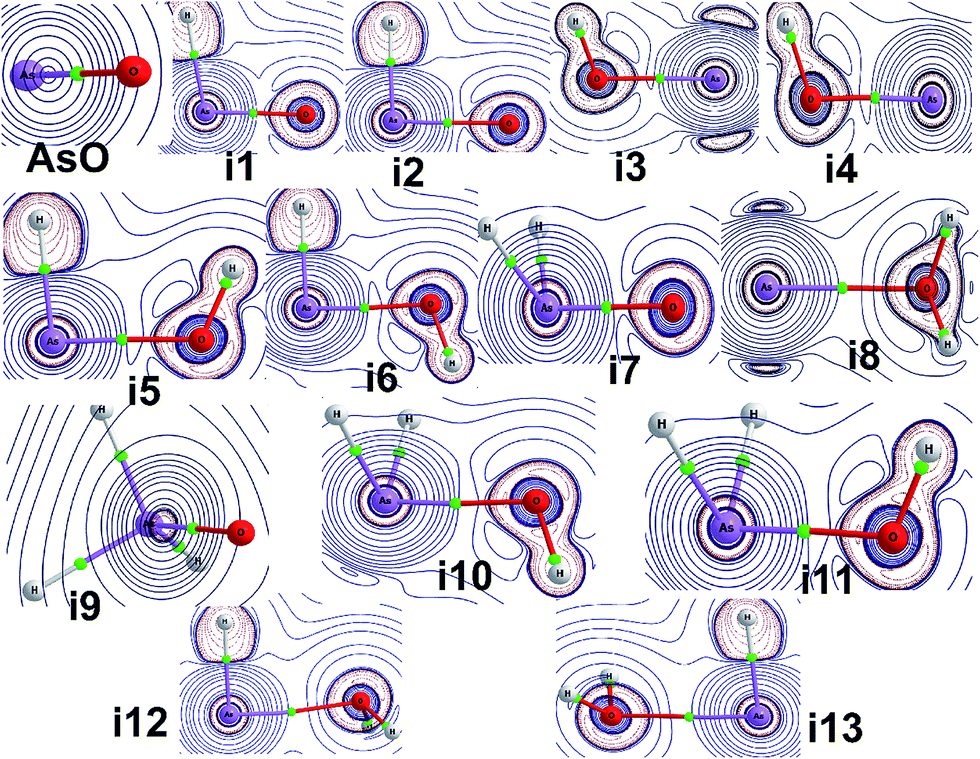 | ||
| Fig. 2 Contour maps of the Laplacian distribution of the electronic charge density [∇2ρ(r)] for the oxoarsine (i1–i4), arsinoyl (i5–i8) and arsine oxide (i9–i13) isomer structures. The blue lines refer to ∇2ρ(r) > 0, and the red lines indicate ∇2ρ(r) < 0. | ||
| Molecule | G4 | G4* | G4MP2 | G4MP2* |
|---|---|---|---|---|
| AsO | 15.9 (14.3) | 15.9 (14.3) | 15.9 (14.3) | 15.9 (14.3) |
| i1 | 8.0 (6.8) | 6.0 (4.8) | 8.1 (6.9) | 6.2 (5.0) |
| i2 | 45.5 (44.4) | 45.8 (44.7) | 46.1 (45.0) | 46.3 (45.3) |
| i3 | 22.6 (21.4) | 22.9 (21.7) | 23.0 (21.8) | 23.3 (22.1) |
| i4 | 5.8 (4.6) | 8.3 (7.1) | 6.0 (4.8) | 8.5 (7.4) |
| i5 | −7.3 (−9.2) | −6.9 (−8.7) | −7.4 (−9.2) | −6.9 (−8.8) |
| i6 | −7.8 (−9.7) | −7.3 (−9.2) | −7.8 (−9.8) | −7.3 (−9.3) |
| i7 | 24.9 (22.9) | 20.9 (18.9) | 25.1 (23.1) | 21.1 (19.1) |
| i8 | 34.7 (32.9) | 39.7 (38.0) | 35.6 (33.8) | 40.6 (38.9) |
| i9 | 8.4 (5.4) | 8.2 (5.2) | 7.8 (4.8) | 7.6 (4.6) |
| i10 | −23.8 (−26.6) | −23.0 (−25.7) | −23.8 (−26.6) | −22.9 (−25.7) |
| i11 | −23.7 (−26.6) | −21.6 (−24.4) | −23.6 (−26.5) | −21.5 (−24.3) |
| i12 | 14.2 (11.8) | 14.9 (12.5) | 14.9 (12.5) | 15.6 (13.2) |
| i13 | 13.9 (11.5) | 14.6 (12.2) | 14.6 (12.2) | 15.3 (12.9) |
3.1. Relative stability, reaction pathways and the heat of formation among AsOHn isomers
The energy profile involving the unimolecular rearrangements among oxoarsine isomers is showed in Fig. 3. The global minimum structure based in the relative energies is i1, which presents a heat of formation that varies from 4.6 to 7.4 kcal mol−1 at 298 K. The energy gap between i1 and i2 is 37.2 kcal mol−1 with CCSD(T)/CBS, while i3 lies 18 kcal mol−1 bellow i1. In this context, it is interesting to see that i4 resides 1.3 kcal mol−1 higher in energy than i1. Additional calculations with QCISD(T, full)/aug-cc-pVQZ showed only a slight difference from the results obtained with CCSD(T)/CBS; the differences were smaller than 3 kcal mol−1. Based on the QCISD(T, full)/aug-cc-pVQZ single-point calculations, there is an estimated energy difference of 2.8 kcal mol−1 between i4 and i1, also confirming that i4 is the second most stable isomer. In addition, important information can also be obtained by comparing these results to the previous theoretical results available in the literature for phosphorous analogous structures, generating a similar picture. For example, Francisco31 predicted the singlet–triplet energy gaps (ΔT–S) for HPO species between 36.5 and 40.1 kcal mol−1, while the ΔS–T values for POH species ranged 15.4–19.3 kcal mol−1 applying CCSD(T) methods. Large values for these energy gaps among HPO isomers were estimated when employing MP2,27 CASSCF28 or even G2,28 while CASPT2 underestimated the CCSD(T) results.28 Values of ΔT–S ranging 24.2–26.1 kcal mol−1 for HPS and a variation of 4.6–11 kcal mol−1 for PSH ΔS–T results were predicted using MP4SDQ, CCSD(T) and CCSD(T)-F12 methodologies.34,40,46 | ||
| Fig. 3 Energy profile (in kcal mol−1) among oxoarsine isomers involving the 1,2-hydrogen shift mechanisms carried out at CCSD(T)/CBS (in parentheses) and QCISD(T, full)/aug-cc-pVQZ (in square brackets) levels with the inclusion of ZPVE (using QCISD/cc-pVTZ values). The pink ball refers to the arsenic atom, while the white and red balls refer to hydrogen and oxygen elements, respectively. | ||
Further insights into the electronic structure of HAsO isomers can be obtained by analyzing unimolecular rearrangement. A barrier height of 53.8 kcal mol−1 is predicted for the i1 → TS1 → i3 proton transfer reaction, and when we looked at this reverse route, there was an estimated energy barrier of 36 kcal mol−1. An inspection on the triplet potential energy surface shows that i2 can isomerize to i4 via a barrier energy of 12 kcal mol−1, with CCSD(T)/CBS, in contrast to the reverse pathway (i4 → TS2 → i2) that can be rearranged by passing over a barrier four times as high. Comparing the refinement in the electronic energies between CCSD(T)/CBS and QCISD(T, full)/aug-cc-pVQZ methods in predicting barrier energies, the differences did not reach 2 kcal mol−1. Compared with phosphorous analogue pathways, the isomerization barrier heights were seen to be between 59.2 and 65.0 kcal mol−1 for the HPO → POH reaction,31 while the singlet and triplet potential energy surface barriers for HPS → PSH rearrangements were 49.8 and 15.3 kcal mol−1, respectively, applying CCSD(T) methods.40 On the other side, arsenic-tin (HAsSn → AsSnH),97 arsenic–silicon (HAsSi → AsSiH)98 and arsenic–germanium (HAsGe → AsGeH)99 1,2-hydrogen shift reactions can occur by smaller energy barriers than those predicted here. Moreover, plotting the singlet and triplet potential energy surfaces obtained by IRC calculations can be seen by an intersystem crossing mechanism where a i1 → i4 pathway is avoided because the singlet potential energy surface can begin to touch the triplet PES at the IRC valley, and then this crossing point region still lies bellow the singlet PES mixing both PES (Fig. S1†). This intersystem crossing region also avoids utilizing the mechanism taking place in the i3 → i4 conversion.
The relative stability that is seen with the G4 and G4MP2 methods failed to predict the global minimum structure among HAsO isomers. G4 and G4MP2 estimate the energy difference between i1 and i4 in −2.2 kcal mol−1, indicating that i4 is the most stable species (see Table S1†). These results are in disagreement with other methodologies, such as CCSD(T)/CBS and QCISD(T, full)/aug-cc-pVQZ that predicts i1 as the global minimum structure, which is in agreement with what is estimated for phosphorous analogous structures.27,28,31,34,40,46 The main reason for these different results is due to a small energy gap among the HAsO isomers and the fact that B3LYP/6-31G(2df,p) (that is used in G4 and G4MP2 methods) underestimates the ZPVE results among these species. The difference between ZPVE results calculated with QCISD/cc-pVTZ and B3LYP/6-31G(2df,p) methods estimates a value of −1.97 kcal mol−1 for i1 and a result of 2.53 kcal mol−1 in the case of i3 (see Table S1†). In contrast, there is an increase of B3LYP accuracy when using the cc-pVTZ basis sets. The efficiency of cc-pVTZ basis sets in the calculation of ZPVE energy can be also confirmed when we look at the mean unassigned error (MUE) and mean absolute error (MAE) in to the prediction of i1 vibrational frequencies (see Table S2†).
Fig. 4 presents the isomerization and H2-release pathways involving the H2AsO isomers. It is also important to mention that QCISD/cc-pVTZ provides a good description of the oxoarsine vibrational frequencies, and, consequently, reliable ZPVE energy values can be seen in the ESI (see Table S4†). The heat of formation at 298 K for i6 varied between −9.76 and −9.20 kcal mol−1, and for i7 ranged 18.91–23.08 kcal mol−1. It is tempting to infer that, although there is only experimental data for i7, it is i6 that is the global minimum structure among H2AsO isomers with an energy gap of 28.8 kcal mol−1 between both species. However, comparing the relative energies between i5 and i6, it is seen that there is a difference lower than 1 kcal mol−1. The largest energy gap is estimated between i6 and i8, with an energy difference of 47.4 kcal mol−1. Further calculations between unrestricted and restricted open-shell wave functions with perturbative-triples correction in the CCSD formalism demonstrated it to be less pronounced in the description of the relative stability among the four isomers (see Table S5†). The differences between UCCSD(T)/aug-cc-pVQZ and ROCCSD(T)/aug-cc-pVQZ are lower than 0.1 kcal mol−1, indicating that the static correlation effects can be well described at an unrestricted formalism at CCSD(T) level. G4 and G4MP2 also provide a reliable description of the relative stability, notwithstanding the energy gap between i6 and i7 is overestimated (see Table S6†). In contrast, G4* and G4MP2* methodologies present similar values with CCSD(T)/CBS in the analysis, whereas the energy difference among these methods is smaller than 0.1 kcal mol−1. Moreover, a similar feature is observed in a comparative analysis of phosphorous analogous radicals, since trans-HPXH (X = O, S) is the global minimum configuration,20,40 as well as in the case of group 15 elements radicals showing a trans-HZYH as the global minimum (Z, Y = N, P, As, Sb, Bi).100–107
 | ||
| Fig. 4 Energy profile (in kcal mol−1) among arsinoyl isomers involving the unimolecular rearrangement routes and the H2-release pathway carried out at CCSD(T)/CBS level (in parentheses) with the inclusion of ZPVE (using QCISD/cc-pVTZ values). The pink ball refers to the arsenic atom, while the white and red balls refer to hydrogen and oxygen elements, respectively. | ||
Considering the reaction pathways of arsinoyl isomers, TS4 refers to the calculated saddle-point structure in the i6 → i5 configuration change process. This conversion occurs by an internal rotation about As–O bond, where the structures with CS symmetries rotate into a saddle-point structure with a C1 point group forming a dihedral angle of 92.8°. TS4 is a less tight transition state structure, with an imaginary frequency of 426 cm−1. This i6 → i5 internal rotation barrier of energy is estimated to be 3.0 kcal mol−1, while TS4 is 2.6 kcal mol−1 above i5. Another reaction path is the i6 → i7 unimolecular rearrangement that converges via the tight TS3 saddle-point structure (−1784 cm−1), where a proton transfer mechanism is seen from the oxygen atom into arsenic. TS3 resides 58.2 kcal mol−1 above i6 and is positioned 27 kcal mol−1 above i7. Furthermore, the i6 → i8 isomerization process occurs by passing through TS5, which is a tight transition state structure with an imaginary frequency of −1709 cm−1. This isomerization mechanism (i6 → i8) involves a high barrier, with TS5 lying 58.5 kcal mol−1 above i6, and the reverse route occurs (i8 → i6) proceeding via an energy barrier of 11 kcal mol−1. Another important mechanism is the H2-release route through TS6 that corresponds to the dehydrogenation of i7, leading to the formation of arsenic monoxide. In TS6, it is estimated that there is an increase of 0.13 Å in the As–H bond length when compared with i7, as well as a decrease of 57.2° and 149° in the HAsH angle and in the dihedral angle, respectively. The H2-elimination path occurs by passing an energy barrier located 28.3 kcal mol−1 above i7, instead of utilizing the mechanism into i7 formation by the AsO + H2 reaction that overcomes an energy barrier of 29.4 kcal mol−1. In this respect, even employing a restricted open-shell wave function with CCSD(T) formalism in these reaction pathways, a difference larger than 1 kcal mol−1 is not predicted when compared with CCSD(T)/CBS.
Fig. 5 shows the energetic profile involving the isomerization and hydrogen release pathways among arsine oxide isomers. It can be noted that i10 is the most stable isomer with a heat of formation at 298 K varying from −25.68 to −26.56 kcal mol−1. The energy difference between i10 and i9 is estimated to be 29.7 kcal mol−1, while i12 resides 43 kcal mol−1 lower in energy than i10. For i9, the heat of formation ranged 4.55–5.42 kcal mol−1. Furthermore, the i10 → i9 rearrangement is undergone via a barrier of 77.2 kcal mol−1, while the reverse reaction also exhibited a sizable barrier of 47.5 kcal mol−1. Examining the results presented by Sola and Toro-Labbe,49 it can be noted that the authors underestimated the barrier heights and overestimated the energy gaps involving arsine oxide and arsinous acid isomerization process, which can happen due to the poor performance method employed in comparative with CCSD(T)/CBS. In the case of the i10 → i12 reaction, the barrier at the corresponding TS12 is calculated to be 55.6 kcal mol−1. Looking at the rotation barriers, it is shown that the hydroxyl rotation barrier height resides in 2 kcal mol−1 considering H2AsOH isomers. On the other hand, the As–H rotation proceeds via a transition state positioned 1 kcal mol−1 above HAs(H2O) isomers. In these arsine oxide isomers, unimolecular decompositions can also occur by eliminating an atomic hydrogen and by H2-release routes. The H2-elimination reaction from i11 proceeds via a tight transition state positioned in a height of 75.5 kcal mol−1 to form i1, which is located in a potential energy well of 38.4 kcal mol−1. The formation of i1 via a H2-release route can also be undergone by i9. The barrier of 49.7 kcal mol−1 with respect to i9 indicates that the i9 → i1 + H2 hydrogen elimination pathways are more favorable when compared with the i11 → i1 + H2 reaction. Alternatively, i9 can undergo a unimolecular decomposition by an atomic hydrogen elimination to form i7 via a loose transition state, TS15, that occurs via a barrier energy of 73 kcal mol−1.
 | ||
| Fig. 5 Energy profile (in kcal mol−1) among arsine oxide isomers involving the unimolecular rearrangement routes and the H2-release pathways carried out at CCSD(T)/CBS level (in parentheses) with the inclusion of ZPVE (using QCISD/cc-pVTZ values). The pink ball refers to the arsenic atom, while the white and red balls refer to hydrogen and oxygen elements, respectively. | ||
3.2. Geometry, electronic properties, vibrational frequencies, bond order indexes, atomic charge distribution, NBO and QTAIM analysis among AsOHn isomers
An important property that can gives some insights into relative stability among the isomer at a molecular orbital level is the second order perturbation energy (E2) that allows the determination of the strength between donor and acceptor NBO. In i1, a large contribution from the donation of the oxygen lone pair electrons (lpO) into the σAs–H anti-bonding orbital ( ) [lpO →
) [lpO →  ] is seen, as well as an equal contribution to the charge transfer from lpO into the arsenic Rydberg orbitals (ryAs) [lpO → rydAs]. Although both of these interactions show a large influence in the stability of i2, the lpO →
] is seen, as well as an equal contribution to the charge transfer from lpO into the arsenic Rydberg orbitals (ryAs) [lpO → rydAs]. Although both of these interactions show a large influence in the stability of i2, the lpO → 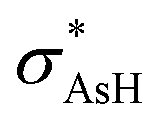 interaction represents a contribution two times higher than lpO → rydAs. A comparison between i1 and i2 shows a decrease of two time in the lpO →
interaction represents a contribution two times higher than lpO → rydAs. A comparison between i1 and i2 shows a decrease of two time in the lpO →  E2 donation energy, while the lpO → rydAs interaction energy decreases four times, which may explain the largest arsenic atomic charge in i1. On the other hand, three important interactions: lpAs →
E2 donation energy, while the lpO → rydAs interaction energy decreases four times, which may explain the largest arsenic atomic charge in i1. On the other hand, three important interactions: lpAs →  lpO → rydAs and σAsO → rydH donations are predicted between AsOH structures. Moreover, the largest energy gap of i2 among their isomers can be also explained by the fact that electrostatic energy plays a major role for this structure. In i3, these three interactions present almost similar contributions to its stability. Nevertheless, the lpO → rydAs charge transfer process is the major contribution for stability of i4, with an E2 energy two times higher than the other two interactions.
lpO → rydAs and σAsO → rydH donations are predicted between AsOH structures. Moreover, the largest energy gap of i2 among their isomers can be also explained by the fact that electrostatic energy plays a major role for this structure. In i3, these three interactions present almost similar contributions to its stability. Nevertheless, the lpO → rydAs charge transfer process is the major contribution for stability of i4, with an E2 energy two times higher than the other two interactions.
Examining the arsenic–oxygen bond nature among HAsO isomers brought important results to light. For example, comparing i1 and i2, a change from a π bond into a σAs–O bond is observed based in the bond order indexes. NRT analysis indicates an i1 reference structure with πAs–O bond (a weight of 98%), which is in good agreement with bond order indexes that show values larger than one. In contrast, an interesting result comes up in the case of i2, where it was seen that there was a resonance weight of 49% for an i2 reference structure presenting a σAs–O bond. However, it is quite noticeable by an analysis of the arsenic–oxygen stretching vibrational frequency [ν(As–O)] that a change in the arsenic–oxygen bond between HAsO species can result in a tremendous red shift of 246 cm−1 from i1 to i2, indicating the change from a π to σAs–O bond that is also reflected by the elongation of the As–O bond (from 1.628 Å in i1 to 1.729 Å in i2) and the decrease in the HAsO angle (from 102° in i1 to 94° in i2).
By analyzing the arsenic–oxygen bond in the AsOH species, it was observed that there was a decrease in the arsenic polarization coefficient by comparison with the i1 value, followed by an increase of the ionic character and a weakening of the As–O bond that consequently lead to a decrease in the values obtained by the different bond order indexes. Looking at the QTAIM results, only a small difference among the several parameters between i3 and i4 was observed. For example, the electronic charge density and the energy density values in the As–O BCP between both species were lower than 4%, and small differences were predicted by examining the ellipticity. Observing the ∇2ρ(r) contour maps in Fig. 2, large differences between the singlet and triplet species cannot be detected. Nevertheless, between i1 and i2 a small difference in the electron charge density localized at oxygen atom can be seen, which can be explained by the slight loss in the electron density. In the i3 ∇2ρ(r) map, it can be seen that there is a very small change in the O–H electron density charge regions that spans over the arsenic charge density region, which is associated with its lone pair electrons. It is also interesting to note that a red shift of only 38 cm−1 is predicted when compared to the ν(As–O) vibrational frequency between i3 and i4. This also mirrors the small change in the bond lengths and bond angles of these two molecules.
Regarding the arsenic–oxygen bond length among the i5–i7 structures, it can be noted that arsinoyl free radical presented the smallest bond distance, 1.679 Å, while trans-hydroxyarsinyl shows a bond length of 1.788 Å. These values are very close to the experimental As–O bond length determined for As(OH)3 (ref. 108) and those for the ionic arsenic–water complexes.109,110 A comparison between the covalent and ionic bond character provided by NRT also suggests a πAs–O bond for i7 due to its large covalent nature and the large polarization on the arsenic atom. There is a similarity in As–O bond length as well as in ν(As–O) vibrational frequencies when comparing i1 and i7 that can be rationalized based on their π bond characters. In i5 and i6, the bond index values are between 0.76 and 1.02 among the three indexes applied here, corresponding to a σAs–O bond. These σAs–O bonds for i5 and i6 are also supported by their ν(As–O) frequency at 700 cm−1. When compared with what has been described in the literature, these ν(As–O) frequency values are similar to those predicted for other systems with an σAs–O bond.111,112 A comparison between i6 and i7 ν(As–O) vibrational modes shows a red shift of 168 cm−1 and a conversion from a σAs–O bond and π bond. Examining the arsenic–oxygen bonds among i5–i7 isomers by NRT noted π and σAs–O bond characters with equal resonance weights for the reference structures, pointing out their large resonance character.
A different picture is observed when considering the As–H2O complex. The As–O bond order in i8 varies from 0.31 to 0.63 among the bond indexes supporting a σAs–O bond nature. This is due to the large As–O bond distance estimated for i8, 2.15 Å. It is worth pointing out that this bond length value is similar to those predicted for aluminum analogues systems.113–116 In addition, a comparison among bond indexes show that the bond order of i8 is two times lower than i6, and the same trend is also seen when comparing their ρ(r) and ∇2ρ(r) results. An analysis of the i8 H(r) result demonstrates the smallest electronic charge concentration between arsenic and oxygen basins, denoting the weakness of the As–O covalent character among all the structures studied here. The largest ellipticity value was also predicted in the i8 As–O bond critical point. This large ellipticity result also highlighted its weak bond nature, which is similar to other weakly bound complexes.117 This is also supported when looking at i8 ν(As–O) frequency, which is the smallest ν(As–O) frequency value among AsOHn isomers, 298 cm−1. In i8, the lpO → lpAs and lpO → rydH charge transfer processes are the main contributions to its stability, but the lpO → lpAs donation is slight higher than the lpO → rydH donation.
Based in the NRT analysis, the i10–i13 reference structure at equilibrium presents a σAs–O bond that is in agreement with the other bond order indexes. Nevertheless, a different picture is seen for i9, where there is a large difference among the bond indexes and the NRT analysis. In i9, the Mayer bond index shows a πAs–O bond, while BDI, bowi and NRT results indicate a σ bond. In contrast, a comparison between i10 and i9 ν(As–O) frequency values shows a red shift of 300 cm−1, which reinforces the i9 πAs–O bond character. This difference among isomer bond characters is also seen when taking a glance at the QTAIM parameters. For example, the electronic charge density in i9 is higher in its arsenic and oxygen basins than in the other isomers. This is also extended to H(r) values, where in i9 the largest results are also seen among arsine oxide isomers, and the difference between i9 and i12 almost reaches an order of magnitude, further supporting the i9 πAs–O bond nature. In this case, it is worth pointing out that the lowest bond orders are predicted for the HAs(H2O) stereoisomers among the three indexes employed. Although the estimated results for i12 and i13 are smaller than those among the bond indexes, NRT reinforces their σAs–O bond characters. When analyzing the population in the arsenic p orbitals at As–O bonds, it can be noted that there is an increase from 90% to 98% in the contribution of p orbitals when comparing i10 and i12 followed by a decrease in the polarization on the arsenic atom. Nevertheless, HAs(H2O) stereoisomers present the largest ellipticity values in their arsenic–oxygen bonds relative to other isomers, indicating their large instability. In addition, analyzing the second order perturbation energy among arsine oxide isomers, it is estimated that the lpO → ν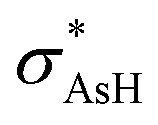 charge transfer process is three times larger than the lpO → rydAs donation, and this difference between both interactions decreases for the arsinous acid stereoisomers. In the case of HAs(H2O) stereoisomers, the largest contribution come from the lpO → rydH charge transfer process.
charge transfer process is three times larger than the lpO → rydAs donation, and this difference between both interactions decreases for the arsinous acid stereoisomers. In the case of HAs(H2O) stereoisomers, the largest contribution come from the lpO → rydH charge transfer process.
To provide information that could shed light on the partial atomic charge distribution, it is important to address that there is a large influence of the population method employed, as can be seen elsewhere.118–121 Therefore, besides QTAIM and NPA, four additional methods were applied, including CHelp,122 CHelpG,123 the Merz–Singh–Kollman method (MK)124 and the Charge Model 5 (CM5).125 In general, the same behavior was described by these four different methods, where a positive charge was predicted for arsenic atoms and a negative charge predicted for oxygen elements. The exceptional cases were those with a weak As–O covalent bond, such as AsOH2 and the HAs(H2O) stereoisomers, where it was estimated that there was a negative formal charge for both elements (see Table S7†).
The charge transfer (δq) between these two atoms [q(As) − q(O)] was also evaluated to understand how the population method influences the δq results among the different As–O bonds. Additional analysis was then carried out in an attempt to find and characterize the linear correlation between the ν(As–O) frequencies and δq. It is important to remember that the previous ν(As–O) results can be attributed to understanding the As–O bond nature (see Table S8†). By correlating the ν(As–O) frequencies calculated with QCISD/cc-pVTZ with the δq results, it can be noted that the correlation increases in the following order: CM5 > NPA > CHelp > MK > CHelpG > QTAIM. These results give a good indicative that δq can work as a tool to describe the As–O bond nature when computed with NPA and CM5 methods. Further, the δq results provided by NPA are larger than those calculated with CM5, suggesting NPA is a better option due to the fact that δq becomes more visible in a visual inspection among a group of few molecules.
Turning to the bond order indexes, it can be observed that unrestricted wave functions require careful analysis before the choice of the bond index, as can be seen for other systems.126,127 This becomes clear when looking at the arsinoyl bond index values (see Table S9†). The Mayer index indicates a π bond nature in H2AsO and the Wiberg index and BDI values orient for a σAs–O bond based in the results, demonstrating a decreasing of 24% when compared with Mayer index. This decrease is also observed among the results obtained with the Wiberg index for the other arsinoyl isomers. Moreover, an attempt to examine the linear correlation between the ν(As–O) frequencies was also performed using the bond order indexes (see Table S10†). This additional analysis was also applied to other indexes as the natural resonance theory (bonrt),86–88 atom–atom overlap natural atomic orbital (bonao)89 and the atom–atom overlap natural localized molecular orbitals/natural population analysis (bonlmo)128 bond order indexes. Therefore, in this case, the linear correlation increased in the following order: bomay > bonao > bowi > bonlmo > BDI > bonrt. Examining bonao and bonlmo, the main negative aspect involving these indexes is the fact that both methods predict very small values, which leads to a small variation among the different bond characters, and, consequently, it becomes a difficult task to characterize the σ/π bond nature for these systems, as was previously seen in literature.127
4. Conclusions
The ab initio study reported herein examined the simplest model systems with an arsenic–oxygen bond. Here was investigated the relative stability among AsOHn isomers (n = 1–3). Among HAsO isomers, the global minimum structure is i1 which presents a heat of formation varying from 4.80 to 6.94 kcal mol−1 at 298 K. The energy gap between i1 and i2 is 37.2 kcal mol−1 with CCSD(T)/CBS, while i4 resides 1.3 kcal mol−1 higher in energy than i1. A barrier height of 54 kcal mol−1 is predicted for the i1 → TS1 → i3 proton transfer reaction, and on the triplet potential energy surface can be seen that i2 can isomerize to i4 proceeding via a barrier energy of 12 kcal mol−1. In the case of H2AsO isomers, there is only experimental data over i7, but it is i6 the global minimum structure with an energy gap of 31 kcal mol−1 between i6 and i7. In this context, the i6 → i5 internal rotation barrier energy is estimated in 3 kcal mol−1, and the i6 → i7 unimolecular rearrangement converges via the tight transition state structure residing 58 kcal mol−1 above i6. Furthermore, analyzing the energetic profile involving the arsine oxide isomers, it can be seen that i10 is the most stable isomer with a heat of formation at 298 K varying from −25.68 to −26.56 kcal mol−1. The i10 → i9 rearrangement can undergo via a barrier of 77 kcal mol−1, while in the case of the i10 → i12 reaction the corresponding barrier height is calculated to be 56 kcal mol−1. A glance at the hydrogen release pathways of arsine oxide isomers shows that the H2-elimination reaction from i11 proceeds via a tight transition state positioned in a height of 75 kcal mol−1 to form i1 which is located in a potential energy well of 38 kcal mol−1. The formation of i1 via a H2-release route can also undergoes by i9. The barrier of 50 kcal mol−1 with respect to i9 indicates that the i9 → i1 + H2 hydrogen elimination pathways is more favorable when compared with the i11 → i1 + H2 reaction.Acknowledgements
This research was supported by CNPq, and the author also wishes to thank CAPES for the research fellowship. This research was supported by resources supplied by the Center for Scientific Computing (NCC/GridUNESP) of the São Paulo State University (UNESP). The author is also grateful to CEPID/CeMEAI (Centro de Pesquisa, Inovação e Difusão em Ciências Matemáticas Aplicadas à Indústria) for the provision of computational facilities.References
- H. Wallmeier and W. Kutzelnigg, J. Am. Chem. Soc., 1979, 101, 2804–2814, DOI:10.1021/ja00505a002.
- M. W. Schmidt, S. Yabushita and M. S. Gordon, J. Phys. Chem., 1984, 88, 382–389, DOI:10.1021/j150647a012.
- W. B. Person, J. S. Kwiatkowski and R. J. Bartlett, J. Mol. Struct., 1987, 157, 237–254, DOI:10.1016/0022-2860(87)87095-3.
- R. Withnall and L. Andrews, J. Phys. Chem., 1987, 91, 784–797, DOI:10.1021/j100288a008.
- J. S. Kwiatkowski, K. Kubulat, W. B. Person, R. J. Bartlett and J. Leszczynski, J. Mol. Struct., 1989, 198, 187–203, DOI:10.1016/0022-2860(89)80038-9.
- J. S. Kwiatkowski and J. Leszczynski, J. Phys. Chem., 1992, 96, 6636–6640, DOI:10.1021/j100195a023.
- J. S. Kwiatkowski and J. Leszczyński, J. Mol. Struct.: THEOCHEM, 1992, 257, 85–103, DOI:10.1016/0166-1280(92)87182-Y.
- I. K. Ahmad, H. Ozeki and S. Saito, J. Chem. Phys., 1999, 110, 912, DOI:10.1063/1.478058.
- Y. Akacem, O. Ouamerali, E. Kassab and E. M. Evleth, J. Phys. Chem., 1992, 96, 1697–1702, DOI:10.1021/j100183a037.
- J. A. Dobado, H. Martinez-Garcia, J. Molina and M. R. Sundberg, J. Am. Chem. Soc., 1998, 120, 8461–8471, DOI:10.1021/ja980141p.
- D. B. Chesnut, J. Am. Chem. Soc., 1998, 120, 10504–10510, DOI:10.1021/ja9822198.
- D. B. Chesnut and A. Savin, J. Am. Chem. Soc., 1999, 121, 2335–2336, DOI:10.1021/ja984314m.
- P. Yin, Z.-L. Wang, Z.-P. Bai, C.-D. Li and X.-Q. Xin, Chem. Phys., 2001, 264, 1–8, DOI:10.1016/S0301-0104(00)00349-9.
- N. L. Haworth and G. B. Bacskay, J. Chem. Phys., 2002, 117, 11175, DOI:10.1063/1.1521760.
- Y.-L. Zhao, J. W. Flora, W. D. Thweatt, S. L. Garrison, C. Gonzalez, K. N. Houk and M. Marquez, Inorg. Chem., 2009, 48, 1223–1231, DOI:10.1021/ic801917a.
- K. Yamada and N. Koga, J. Comput. Chem., 2013, 34, 149–161, DOI:10.1002/jcc.23118.
- G. Manca, M. Caporali, A. Lenco, M. Peruzzini and C. Mealli, J. Organomet. Chem., 2014, 760, 177–185, DOI:10.1016/j.jorganchem.2013.10.043.
- S. Yabushita and M. S. Gordon, Chem. Phys. Lett., 1985, 117, 321–325, DOI:10.1016/0009-2614(85)85236-2.
- R. Withnall and L. Andrews, J. Phys. Chem., 1988, 92, 4610–4619, DOI:10.1021/j100327a012.
- M. T. Nguyen and T.-K. Ha, Chem. Phys., 1989, 131, 245–253, DOI:10.1016/0301-0104(89)80173-9.
- S. P. So, J. Phys. Chem., 1990, 94, 2344–2347, DOI:10.1021/j100369a028.
- T. Hirao, S. Saito and H. Ozeki, J. Chem. Phys., 1996, 105, 3450, DOI:10.1063/1.472534.
- S. S. Wesolowski, E. M. Johnson, M. L. Leininger, T. D. Crawford and H. F. Schaefers, J. Chem. Phys., 1998, 109, 2694, DOI:10.1063/1.476869.
- M. A. Gharaibeh, D. J. Clouthier and R. Tarroni, J. Chem. Phys., 2011, 135, 214307, DOI:10.1063/1.3664903.
- M. Larzilliere and M. E. Jacox, J. Mol. Spectrosc., 1980, 79, 132–150, DOI:10.1016/0022-2852(80)90298-2.
- S. Saito, Y. Endo and E. Hirota, J. Chem. Phys., 1986, 84, 1157, DOI:10.1063/1.450505.
- P. Redondo, A. Largo, C. Barrientos and J. M. Ugalde, J. Phys. Chem., 1991, 95, 4318–4323, DOI:10.1021/j100164a027.
- A. Luna, M. Merchan and B. O. Ross, Chem. Phys., 1995, 196, 437–445, DOI:10.1016/0301-0104(95)00092-3.
- B. S. Tackett and D. J. Clouthier, J. Chem. Phys., 2002, 117, 10604, DOI:10.1063/1.1521130.
- H. Ozeki and S. Saito, J. Mol. Spectrosc., 2003, 219, 305–312, DOI:10.1016/S0022-2852(03)00052-3.
- J. S. Francisco, J. Chem. Phys., 2003, 287, 303–316, DOI:10.1016/S0301-0104(02)00972-2.
- C. Puzzarini, J. Mol. Struct., 2006, 780–781, 238–246, DOI:10.1016/j.molstruc.2005.06.042.
- E. P. F. Lee, D. K. W. Mok, F.-T. Chau and J. M. Dyke, J. Chem. Phys., 2007, 127, 214305, DOI:10.1063/1.2790892.
- M. T. Nguyen, Chem. Phys., 1987, 117, 91–99, DOI:10.1016/0301-0104(87)80099-X.
- P. V. Sudhakar and K. Lammertsma, J. Org. Chem., 1991, 56, 6067–6071, DOI:10.1021/jo00021a021.
- Z. Mielke and L. Andrews, J. Phys. Chem., 1993, 97, 4313–4319, DOI:10.1021/j100119a011.
- C. J. Cramer, Chem. Phys. Lett., 1993, 202, 297–302, DOI:10.1016/0009-2614(93)85281-R.
- W. N. Wang, W. L. Wang, Q. Luo and Q. S. Li, Chem. Phys. Lett., 2005, 415, 370–374, DOI:10.1016/j.cplett.2005.09.016.
- P. Yin, C. Wang, H. Zheng, G. Yin and T. Zhou, J. Chem. Res., 2006, 5, 303–305, DOI:10.3184/030823406777410963.
- R. B. Viana and A. S. Pimentel, J. Chem. Phys., 2007, 127, 204306, DOI:10.1063/1.2800012.
- J. Yang, X. Zhang, D. J. Clouthier and R. Tarroni, J. Chem. Phys., 2009, 131, 204307, DOI:10.1063/1.3266944.
- J. Yang, D. J. Clouthier and R. Tarroni, J. Chem. Phys., 2009, 131, 224311, DOI:10.1063/1.3270160.
- R. A. Grimminger, D. J. Clouthier and R. Tarroni, J. Chem. Phys., 2011, 135, 214306, DOI:10.1063/1.3662416.
- D. T. Halfen, D. J. Clouthier, L. M. Ziurys, V. Lattanzi, M. C. McCarthy, P. Thaddeus and S. Thorwirth, J. Chem. Phys., 2011, 134, 134302, DOI:10.1063/1.3562374.
- R. Grimminger, D. J. Clouthier, R. Tarroni, Z. Wang and T. J. Sears, J. Chem. Phys., 2013, 139, 174306, DOI:10.1063/1.4827099.
- S. B. Yaghlane, C. E. Cotton, J. S. Francisco, R. Linguerri and M. Hochlaf, J. Chem. Phys., 2013, 139, 174313, DOI:10.1063/1.4827520.
- L. Andrews, R. Withnall and B. W. Moores, J. Phys. Chem., 1989, 93, 1279–1285, DOI:10.1021/j100341a022.
- W. Schneider, W. Thiel and A. Komornicki, J. Phys. Chem., 1990, 94, 2810–2814, DOI:10.1021/j100370a017.
- M. Sola and A. Toro-Labbe, J. Phys. Chem. A, 1999, 103, 8847–8852, DOI:10.1021/jp990576e.
- M. T. Monahan-Pendergast, M. Przybylek, M. Lindblad and J. Wilcox, Atmos. Environ., 2008, 42, 2349–2357, DOI:10.1016/j.atmosenv.2007.12.028.
- R. Tarroni and D. J. Clouthier, J. Chem. Phys., 2009, 131, 114310, DOI:10.1063/1.3230132.
- S.-G. He, F. X. Sunahori, J. Yang and D. J. Clouthier, J. Chem. Phys., 2009, 131, 114311, DOI:10.1063/1.3230142.
- R. Grimminger and D. J. Clouthier, J. Chem. Phys., 2011, 135, 184308, DOI:10.1063/1.3658645.
- A. Orthaber, A. F. Sax and K. A. Francesconi, J. Comput. Chem., 2012, 33, 112–117, DOI:10.1002/jcc.21950.
- L. Stojanovic, J. Phys. Chem. A, 2012, 116, 8624–8633, DOI:10.1021/jp304786r.
- J. Morris, H. Bui and B. S. Ault, Chem. Phys., 2008, 348, 203–208, DOI:10.1016/j.chemphys.2008.03.013.
- S. Aldridge and A. J. Downs, Chem. Rev., 2001, 101, 3305–3366, DOI:10.1021/cr960151d.
- J. Gauss and D. Cremer, Chem. Phys. Lett., 1988, 150, 280–286, DOI:10.1016/0009-2614(88)80042-3.
- E. A. Salter, G. W. Trucks and R. J. Bartlett, J. Chem. Phys., 1989, 90, 1752–1766, DOI:10.1063/1.456069.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806, DOI:10.1063/1.462569.
- A. K. Wilson, D. E. Woon, K. A. Peterson and T. H. Dunning Jr, J. Chem. Phys., 1999, 110, 7667–7676, DOI:10.1063/1.478678.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789, DOI:10.1103/PhysRevB.37.785.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627, DOI:10.1021/j100096a001.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924, DOI:10.1063/1.1724823.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61–69, DOI:10.1021/ct0499783.
- T. J. Lee and P. R. Taylor, Int. J. Quantum Chem., 1989,(S23), 199–207, DOI:10.1002/qua.560360824.
- T. J. Lee, A. P. Rendell and P. R. Taylor, J. Phys. Chem., 1990, 94, 5463–5568, DOI:10.1021/j100377a008.
- J. C. Rienstra-Kiracofe, W. D. Allen and H. F. Schaefer III, J. Phys. Chem. A, 2000, 104, 9823–9840, DOI:10.1021/jp001041k.
- X. Yang, A. W. Jasper, B. R. Giri, J. H. Kiefer and R. S. Tranter, Phys. Chem. Chem. Phys., 2011, 13, 3686–3700, 10.1039/C0CP01541E.
- L. B. Harding, S. J. Klippenstein and A. W. Jasper, Phys. Chem. Chem. Phys., 2007, 9, 4055–4070, 10.1039/B705390H.
- K. A. Peterson, D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1994, 100, 7410–7415, DOI:10.1063/1.466884.
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975, DOI:10.1063/1.453520.
- A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243–252, DOI:10.1016/S0009-2614(98)00111-0.
- B. A. Hess, Phys. Rev. A, 1986, 33, 3742–3748, DOI:10.1103/PhysRevA.33.3742.
- B. A. Hess, Phys. Rev. A, 1985, 32, 756–763, DOI:10.1103/PhysRevA.32.756.
- W. A. de Jong, R. J. Harrison and D. A. Dixon, J. Chem. Phys., 2001, 114, 48, DOI:10.1063/1.1329891.
- K. Raghavachari, J. A. Pople, E. S. Replogle and M. Head-Gordon, J. Phys. Chem., 1990, 94, 5579–5586, DOI:10.1021/j100377a033.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- T. A. Keith, AIMAll (Version 11.12.19), TK Gristmill Software, Overland Park KS, USA, 2011, http://aim.tkgristmill.com Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 6.0, Theoretical chemistry institute, University of Wisconsin, Madison, WI, 2013, http://nbo6.chem.wisc.edu/ Search PubMed.
- D. Cremer and E. Kraka, Angew. Chem., 1984, 23, 627–628, DOI:10.1002/anie.198406271.
- X. Fradera, M. A. Austen and R. F. W. Bader, J. Phys. Chem. A, 1999, 103, 304–314, DOI:10.1021/jp983362q.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096, DOI:10.1016/0040-4020(68)88057-3.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274, DOI:10.1016/0009-2614(83)80005-0.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593–609, DOI:10.1002/(SICI)1096-987X(19980430)19.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 610–627, DOI:10.1002/(SICI)1096-987X(19980430)19.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 628–646, DOI:10.1002/(SICI)1096-987X(19980430)19.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746, DOI:10.1063/1.449486.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108, DOI:10.1063/1.2436888.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 127, 124105, DOI:10.1063/1.2770701.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern and J. A. Pople, J. Chem. Phys., 1997, 106, 1063, DOI:10.1063/1.473182.
- M. W. Chase Jr, NIST-JANAF Thermochemical Tables, J. Phys. Chem. Ref. Data, Monograph, 4th edn, 1998, vol. 9. p. 1 Search PubMed.
- D. K. W. Mok, E. P. F. Lee, F.-T. Chau and J. M. Dyke, Phys. Chem. Chem. Phys., 2011, 13, 9540–9553, 10.1039/c1cp20490d.
- R. B. Viana and A. B. F. da Silva, J. Mol. Model., 2014, 20, 2372, DOI:10.1007/s00894-014-2372-8.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y.-H. Hu and M.-D. Su, J. Phys. Chem. A, 2003, 107, 4130–4135, DOI:10.1021/jp022236q.
- C.-H. Lai, M.-D. Su and S.-Y. Chu, Organometallics, 2002, 21, 397–400, DOI:10.1021/om010792l.
- C.-H. Lai, M.-D. Su and S.-Y. Chu, Polyhedron, 2002, 21, 579–585, DOI:10.1016/S0277-5387(01)01021-X.
- W. W. Schoeller, C. Begemann, U. Tubbesing and J. Strutwolf, J. Chem. Soc., Faraday Trans., 1997, 93, 2957–2962, 10.1039/A701636K.
- M.-D. Su, C.-H. Lai, M.-J. Cheng and S.-Y. Chu, J. Phys. Chem. A, 2004, 108, 8448–8455, DOI:10.1021/jp0402636.
- K. Szopa, M. Musiał and S. A. Kucharski, Int. J. Quantum Chem., 2008, 108, 2108–2116, DOI:10.1002/qua.21718.
- C.-H. Lai and M.-D. Su, J. Comput. Chem., 2008, 29, 2487, DOI:10.1002/jcc.20970.
- T. Lu, A. C. Simmonett, F. A. Evangelista, Y. Yamaguchi and H. F. Schaefer III, J. Phys. Chem. A, 2009, 113, 13227–13236, DOI:10.1021/jp904028a.
- R. B. Viana, P. S. S. Pereira, L. G. M. Macedo and A. S. Pimentel, Chem. Phys., 2009, 363, 49–58, DOI:10.1016/j.chemphys.2009.07.008.
- S. Vogt-Geisse and H. F. Schaefer III, J. Chem. Theory Comput., 2012, 8, 1663–1670, DOI:10.1021/ct300221e.
- C.-H. Lai and M.-D. Su, J. Organomet. Chem., 2014, 751, 379, DOI:10.1016/j.jorganchem.2013.07.021.
- A. Ramírez-Solís, R. Mukopadhyay, B. P. Rosen and T. L. Stemmler, Inorg. Chem., 2004, 43, 2954–2959, DOI:10.1021/ic0351592.
- S. Bagchi, I. Bhattacharyya, B. Mondal and A. K. Das, Mol. Phys., 2011, 109, 933–941, DOI:10.1080/00268976.2011.558857.
- S. Bagchi and A. K. Das, Chem. Phys. Lett., 2011, 508, 191–196, DOI:10.1016/j.cplett.2011.04.045.
- A. Adamescu, H. Gray, K. M. E. Stewart, I. P. Hamilton and H. A. Al-Abadleh, Can. J. Chem., 2010, 88, 65–77, DOI:10.1139/V09-147.
- A. Bhattacharjee, A. B. Pribil, L. H. V. Lim, T. S. Hofer, B. R. Randolf and B. M. Rode, J. Phys. Chem. B, 2010, 114, 3921–3926, DOI:10.1021/jp911860y.
- S. Alvarez-Barcia and J. R. Flores, Chem. Phys. Lett., 2009, 470, 196–202, DOI:10.1016/j.cplett.2009.01.069.
- S. Alvarez-Barcia and J. R. Flores, J. Chem. Phys., 2009, 131, 174307, DOI:10.1063/1.3253049.
- S. Alvarez-Barcia and J. R. Flores, Chem. Phys., 2010, 374, 131–137, DOI:10.1016/j.chemphys.2010.07.013.
- S. Alvarez-Barcia, J. R. Flores, G. Granucci and M. Persico, J. Phys. Chem. A, 2013, 117, 67–74, DOI:10.1021/jp310034c.
- R. B. Viana and A. B. F. da Silva, Polyhedron, 2015, 89, 160–167, DOI:10.1016/j.poly.2015.01.010.
- F. Martin and H. Zipse, J. Comput. Chem., 2005, 26, 97–105, DOI:10.1002/jcc.20157.
- A. D. Kulkarni and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 2325–2332, DOI:10.1021/ct200188n.
- R. B. Viana, E. D. A. Santos, L. J. Valencia, R. M. Cavalcante, E. B. Costa, R. Moreno-Fuquen and A. B. F. da Silva, Spectrochim. Acta, Part A, 2013, 102, 386–392, DOI:10.1016/j.saa.2012.09.094.
- R. B. Viana, G. L. O. Ribeiro, S. F. F. Santos, D. E. Quintero, A. B. Viana, A. B. F. da Silva and R. Moreno-Fuquen, Spectrochim. Acta, Part A, 2016, 162, 16–26, DOI:10.1016/j.saa.2016.02.037.
- L. E. Chirlian and M. M. Francl, J. Comput. Chem., 1987, 8, 894–905, DOI:10.1002/jcc.540080616.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373, DOI:10.1002/jcc.540110311.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145, DOI:10.1002/jcc.540050204.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 527–541, DOI:10.1021/ct200866d.
- P. Szarek, Y. Sueda and A. Tachibana, J. Chem. Phys., 2008, 129, 094102, DOI:10.1063/1.2973634.
- R. B. Viana, A. R. Guimaraes, A. R. de Souza and A. B. F. da Silva, J. Mol. Model., 2014, 20, 2074, DOI:10.1007/s00894-014-2074-2.
- A. E. Reed and P. V. R. Schleyer, Inorg. Chem., 1988, 27, 3969–3987, DOI:10.1021/ic00295a018.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra09517h |
| This journal is © The Royal Society of Chemistry 2016 |