
Mechanism aspects of the hydrogenation of acrylonitrile on Ni and Pd surfaces†
Xin Geab,
Jiongbin Pana,
Xinzhi Chena,
Chao Qian*a and
Shaodong Zhou*c
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou, P. R. China. E-mail: qianchao@zju.edu.cn
bSchool of Chemical and Material Engineering, Jiangnan University, Wuxi, P. R. China
cInstitut für Chemie, Technische Universität Berlin, Berlin, Germany. E-mail: shaodong.zhou@campus.tu-berlin.de
First published on 25th May 2016
Abstract
A combined experimental and theoretical investigation on the hydrogenation of acrylonitrile catalyzed by Ni and Pd is presented. The experiments were conducted in an autoclave and monitored by a computer remote, while the Ni(111) and Pd(111) surface models were employed for theoretical study using density functional theory. Acrylonitrile is preferentially adsorbed on both surfaces through all atoms of the molecular backbone interacting with metal surfaces. For the hydrogenation of the vinyl moiety on Ni, the preferred pathway is initiated by formation of the C1–H bond, while on the Pd surface this step is alternated by making a C2–H bond. Comparison of the reaction pathways for Ni(111) and Pd(111) indicates lower energy barriers for further hydrogenation of the nitrile group on the latter, thus in line with the experimentally observed higher selectivity of the hydrogenation of vinyl catalyzed by Ni.
Introduction
Propionitrile is the most simple unsaturated aliphatic nitrile, which is widely used as a solvent or precursor for preparing more valuable compounds.1 In addition to the ammoxidation of propanol,2 catalytic hydrogenation of acrylonitrile is an alternative approach to produce propionitrile. However, as acrylonitrile possesses both a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and a C
C and a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond,3 the hydrogenations of both the vinyl and the nitrile groups are possible. The transition metal, e.g. Ru,4 Ni5 and Pd,6 was used to catalyze the hydrogenation of acrylonitrile to propionitrile, while propylamine is inevitably generated as the byproduct. Previous experimental evidences suggest that the reaction proceeds sequentially by generating propionitrile first, and further hydrogenation to propylamine afterward.5a
N bond,3 the hydrogenations of both the vinyl and the nitrile groups are possible. The transition metal, e.g. Ru,4 Ni5 and Pd,6 was used to catalyze the hydrogenation of acrylonitrile to propionitrile, while propylamine is inevitably generated as the byproduct. Previous experimental evidences suggest that the reaction proceeds sequentially by generating propionitrile first, and further hydrogenation to propylamine afterward.5a
Although there exist quite some experimental studies on the hydrogenation of acrylonitrile catalyzed by transition metals, only a few of them focus on the reaction mechanisms and kinetics. Deng et al. proposed three reaction pathways for the hydrogenation of acrylonitrile catalyzed by Ni–B/SiO2,5e–g corresponding to the hydrogenations of the CC bond, the CN bond, and both the two, respectively. They elucidated that the electronic interaction between Ni and B in Ni–B alloy improve the selectivity of hydrogenation of the CC bond. Due to the partial electron transfer from B to Ni, the electron-enriched Ni could effectively inhibit the adsorption of nitrile group.
While the adsorption of acrylonitrile on the transition metal surfaces, e.g. Cu,7 Ni,8 Pt and Au,3 has attracted much attention, reports on the hydrogenation mechanisms are very rare. Once molecules are adsorbed on metal surface, their structural change have important effect on their reactivity.7f The conjugation between the π orbitals of the CC and CN bonds lowers the electronic density of CC bond via the delocalizing the CC bond π orbital to the whole molecule.9 Thus, if the reactivity of CC bond group in the interaction is strengthen compared with the nitrile group, the selectivity of the hydrogenation will be improved.10 Parent et al.3 studied the interaction of acrylonitrile on Pt(111) and Au(111) by means of near-edge X-ray fine structure (NEXAFS); they found that acrylonitrile was physisorbed on Au(111) and chemisorbed on Pt(111). Fredriksson et al.8a investigated the interaction of several metals with acrylonitrile both experimentally and theoretically, and it was found that the acrylonitrile could form π–d bonds with Ni and Cu atoms. Geskin et al.7b described the acrylonitrile adsorbed on Cu(100) via coordination to two copper atoms with vinyl and nitrile ends; further, Crispin et al.7c reported the similar result, which was confirmed experimentally by X-ray Photoelectron Spectroscopy. Some recent theoretical study by performing periodic boundary condition,7e–g were consistent with the previous results.7b,c For acrylonitrile adsorbed on Ni(100), Crispin et al.8b found that all atoms of acrylonitrile form covalent bonds with five nickel atoms, respectively. The adsorption type on Ni(100) was the formation of π–d bond, which is different from the σ–d adsorption on Cu(100).
In this paper, performances of different transition metal catalysts in the hydrogenation of acrylonitrile were examined. The detailed adsorption and reaction mechanisms on Ni(111) and Pd(111) surfaces were investigated using density functional theory (DFT) calculations.
Computational details
DFT calculations were carried out by using the CASTEP program package in Materials Studio of Accelrys Inc,11 where the Perdew, Burke, Erzenhof gradient corrected functional (GGA-PBE) is chosen together with plane wave basis functions with spin polarization.12 The linear and quadratic synchronoustransit (LST/QST) complete search was chose to search for transition state of the reaction.13 The simulation of core electron was performed by Ultrasoft pseudopotential (USP).14 In order to improve computational performance, energy cut-off was set 400.0 eV.Ni(111) and Pd(111) surfaces were modeled by using a three-layer periodic slab model with a (5 × 5) super cell. Then by building a 10 Å vacuum slab, the adsorption and reaction occurs in this cell. The reciprocal space of the (5 × 5) super cell was sampled using the 3 × 3 × 1 k-points grid. Larger k-points sets were needed if more accurate energy value wanted. Study in this work focused on the relative results of different systems, and the k-points set of (3 × 3 × 1) was selected to obtain qualitative results. For the geometry optimization, all Ni and Pd atoms were constrained except the uppermost layer, with setting the convergence tolerances of energy and displacement to 2 × 10−5 eV per atom and 2 × 10−3 Å, respectively, and setting the SCF tolerance to 2 × 10−6 eV per atom.
Chemisorption energies were calculated using the following formulas:
| ΔEads = Eadsorbate–metal − Eadsorbate − Emetal |
For a hydrogenation reaction, such as R + H → RH, the energy barrier was calculated as follows:
| ΔEReact = ETs − ER–metal |
Result and discussion
We have screened several transition metals, e.g. Ni, Pd, Co, Cu, and Pt, as the active component for the hydrogenation of acrylonitrile.15 It has been found that RANEY® Ni catalyzed hydrogenation of acrylonitrile proceeds fast with excellent selectivity. In contrast, Pd/C (5 wt%) exhibits less activity and poorer selectivity, producing propylamine and dipropylamine as byproducts (see the ESI†). In this paper, we attempted to study the hydrogenation of acrylonitrile at a molecule level, so as to reveal the cause for the different reactivities and selectivities for the reactions promoted by Ni and Pd/C.The FT-IR spectra of RANEY® Ni catalyst recovered after the catalytic hydrogenation was obtained (in Fig. 1). The spectrum exhibited broad peaks at around 3425 cm−1, which can be attributed to stretching vibration of the amine group. The presence of the peak at 1631 cm−1 is due to the CN stretching vibration. The bending vibration of methyl group led to the peak around 1384 cm−1. Thus, appearance of the peak at 1631 cm−1 implies that the intermediate may be imine when the nitrile was reduced to amine group. In the further hydrogenation, some byproducts e.g. propylamine, dipropylamine, 3-(propylamino)propanenitrile, 3-(dipropylamino)propanenitrile, were obtained by MS (see Fig. S2–S5 in ESI†). Therefore, the possible pathway was proposed as shown in Fig. 2. The further hydrogenation of acrylonitrile led to the appearance of the propylamine. Other byproducts such as 3-(propylamino)propanenitrile, 3-(dipropylamino)propanenitrile, were formed by the addition and disproportionation, which were confirmed in MS. By compared with RANEY® Ni with Pd/C, we attempted to answer the fundamental question: how metal catalyst influences the hydrogenation of acrylonitrile?
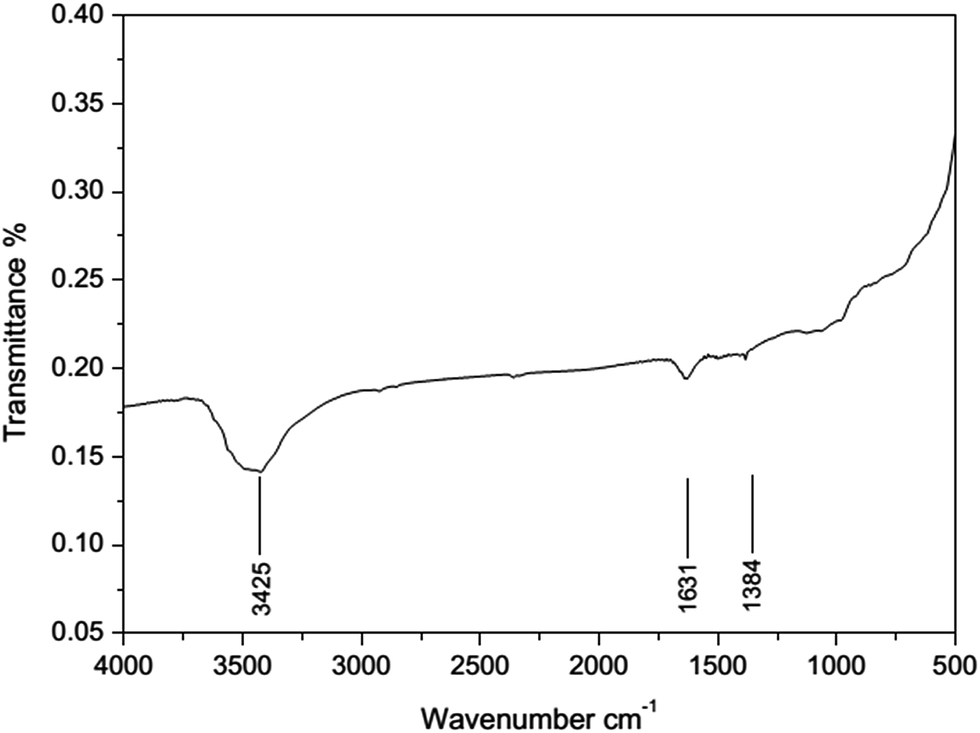 | ||
| Fig. 1 FI-IR spectrum of recovered RANEY® Ni in the hydrogenation of acrylonitrile. | ||
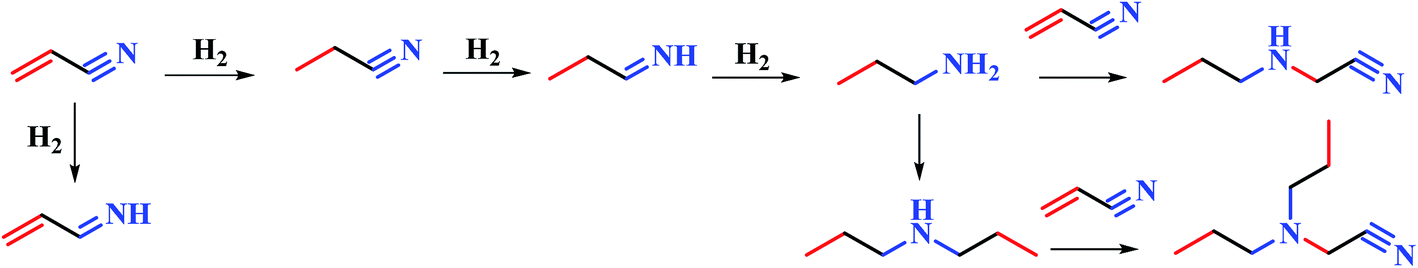 | ||
| Fig. 2 The proposed mechanism of hydrogenation of acrylonitrile. | ||
As shown in Fig. 3, RANEY® Ni catalyst has three diffraction peaks of Ni(111), Ni(200) and Ni(220). Meanwhile, although the strongest peak of Pd/C catalyst is attributed to the activated carbon, it is found obviously that Pd/C catalyst has two diffraction peaks of Pd(111) and Pd(110). Moreover, Ni(111) and Pd(111) are the main diffraction peak of RANEY® Ni and Pd/C, respectively. Also, surface chemisorption of kinds of molecules on Ni(111) and Pd(111) have been studied, such as acetylene,16 HCN17 and butadiene.18 In addition, as Ni(111) and Pd(111) are congener model system,19 they were used in comparative studies to reveal the reaction mechanism and the influence of metal catalysts.
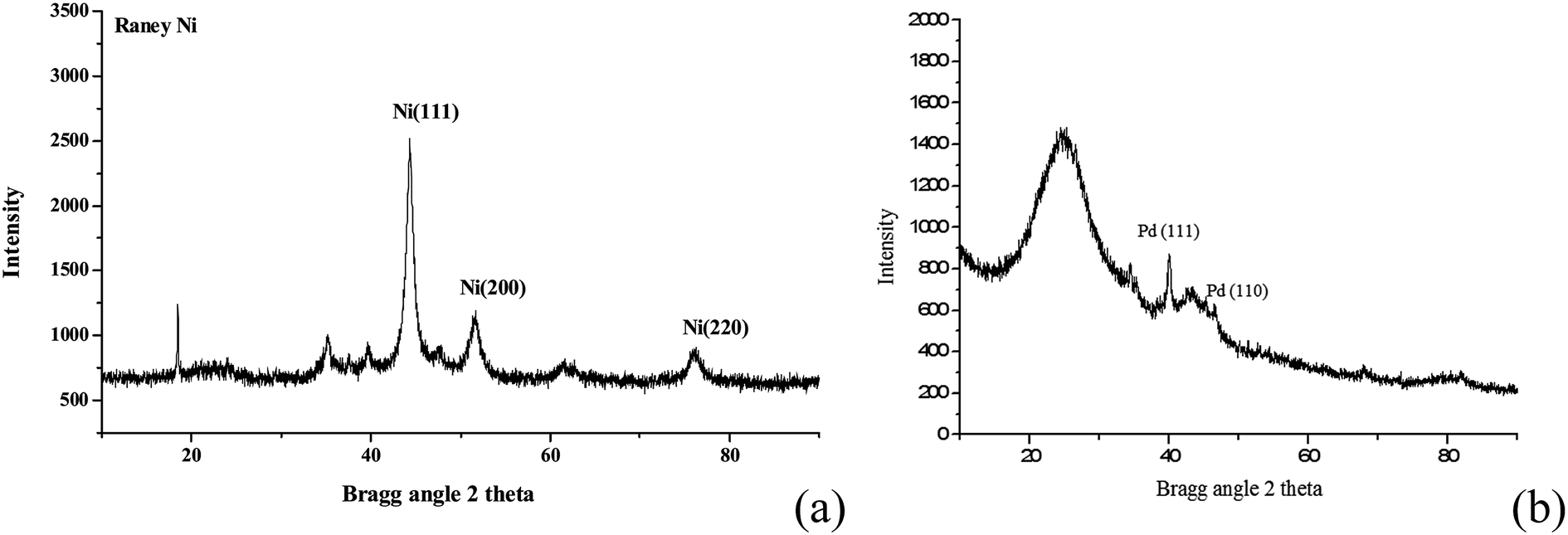 | ||
| Fig. 3 X-ray diffraction patterns of RANEY® Ni (a) and Pd/C (b) catalysts. | ||
Adsorption of acrylonitrile on Ni(111) and Pd(111)
For bare acrylonitrile in gas phase, the calculated bond lengths and angels are 1.344 Å (1.339 Å) for C1–C2, 1.427 Å (1.426 Å) for C2–C3, 1.183 Å (1.164 Å) for C3–N and 178.599° (180°) for C2–C3–N, respectively, which are consistent with previous experimental results.7bBy constructing and optimizing several initial configurations of acrylonitrile on Ni(111) and Pd(111) surfaces, three stable configurations were found for the bridge-, hollow- and top-adsorptions on the metal surfaces, respectively; more structural information is provided in the ESI.† Among these adsorption modes, N-fcc is found to be most stable one for both Ni(111) and Pd(111), and it is thus used for subsequent investigation on reaction mechanisms. As shown in Fig. 4a, acrylonitrile is adsorbed on on the Ni(111) surface via a di-π mode (N-fcc-C3-hcp), which is similar with the previously reported adsorption type on the Ni(100) surface.8b Thus, all 2p-π molecular orbitals interact with d and s atomic orbitals of metals to form new bonds. Accordingly, the C1–C2, C2–C3 and C3–N bond are elongated by 0.122, 0.024 and 0.117 Å, respectively. The bond lengths of N–Ni are 1.940 and 2.004 Å, and the other C–Ni bond lengths were 1.875–2.135 Å. For Pd(111), the adsorption is defined as the N-fcc-C3-fcc mode, which is slightly different from the N-fcc-C3-hcp, that is, only four palladium atoms bond with the acrylonitrile molecule. On Pd(111), the C1–C2, C2–C3 and C3–N bond are elongated by 0.115, 0.036 and 0.065 Å, respectively. The bond lengths of N–Pd is 2.095 Å, and the other C–Ni bond lengths are 2.141–2.167 Å. The slightly longer bond lengths of C-metal and N-metal implies weaker adsorption of acrylonitrile on Pd as compared to that on Ni. This is further proved by adsorption enthalpies, which are −154.8 kJ mol−1 and −148.12 kJ mol−1 on Ni(111) and Pd(111), respectively.
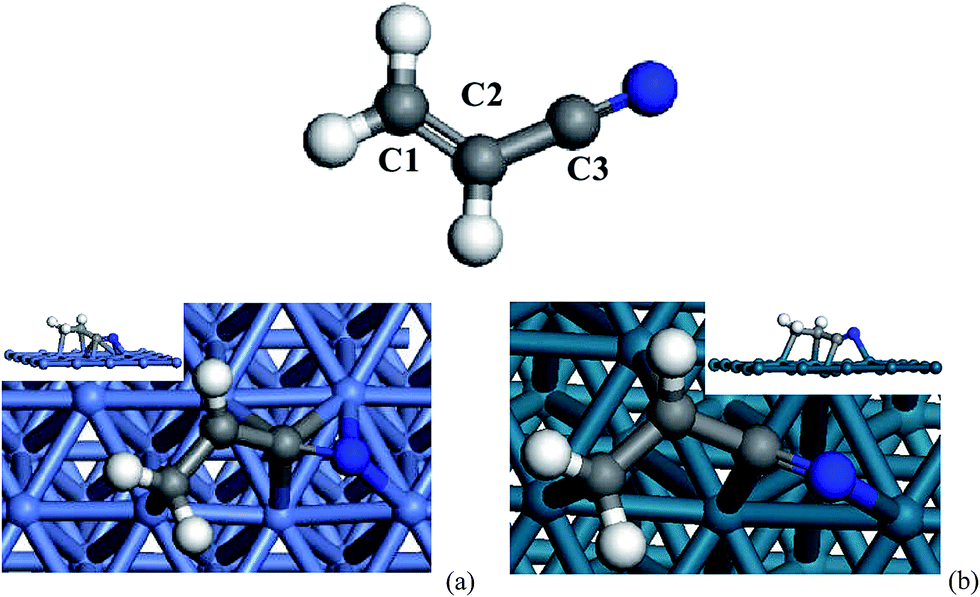 | ||
| Fig. 4 The most stable site of acrylonitrile adsorbed on Ni(111) (a) and Pd(111) (b). | ||
Hydrogenation of acrylonitrile to propionitrile on Ni(111) and Pd(111)
Having found the stable adsorption mode, the hydrogenation of acrylonitrile on Ni(111) and Pd(111) was studied. Initiated by formation of the C1–H and C2–H bonds, two possible pathways were located, i.e. path A and B; associated potential energy surfaces (PESs) are shown in Fig. 5 (see Fig. S7–S8 in ESI†). Prior to the hydrogenation, the hydrogen molecule is dissociated to two H atoms adsorbed on the top site of metal surfaces barrierlessly.20 The dissociated H atom is migrated to the hollow site from the top site.19b,21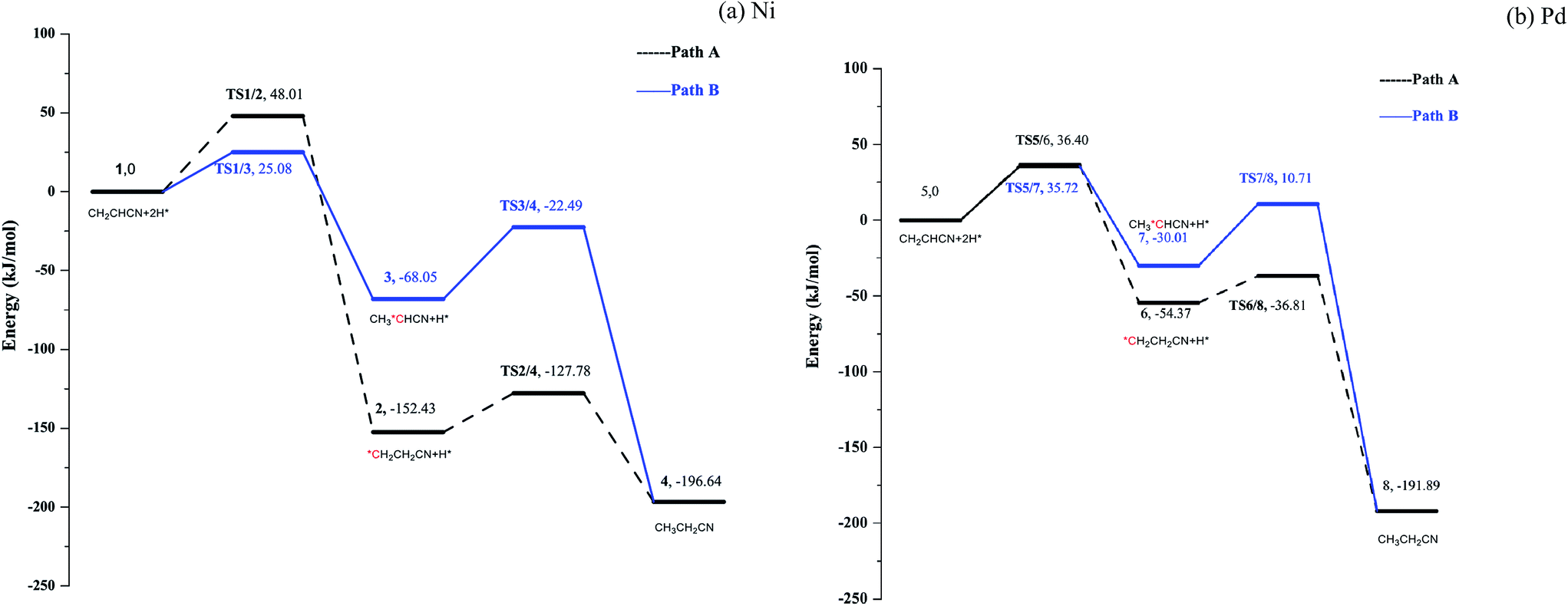 | ||
| Fig. 5 Potential energy profiles of hydrogenation of acrylonitrile to propionitrile on Ni(111) (a) and Pd(111) (b). | ||
For path A, the C1–H bonds are formed upon breaking the C1-metal bonds by surmounting barriers of 48.01 and 36.40 kJ mol−1 for Ni and Pd, respectively. Subsequently, the other intermediates TS2/4 and TS6/8 are obtained by making the C2–H bonds, with energy barriers of 24.65 and 17.56 kJ mol−1 for Ni and Pd, respectively.
For path B, the initial formation of C2–H bonds is accompanied by barriers of 25.08 and 35.72 kJ mol−1 for Ni and Pd, respectively. On Ni(111) surface, upon formation of the C2–H bond, the C1 atom slightly moves away from the Ni surface. It prolongs the length of C1–Ni bond from 1.967 Å to 1.979 Å, which makes it easier to break the C1–Ni bond and lowers the energy barrier for formation of the C1–H bond in path B (45.56 kJ mol−1). On Pd(111), formation of the C2–H stretch the C1 atom close to the metal surface, and cleavage of the C1–Pd bond is thus more difficult in path B (with a barrier of 40.72 kJ mol−1) as compared to that in path A. Thus, on Ni, path A is a preferred hydrogenation pathway, while path B is more favorable for Pd. However, the reactions on Ni and Pd surfaces have one feature in common: formation of the C1–H bond corresponds to the rate-limiting step.
Further hydrogenation on Ni(111) and Pd(111)
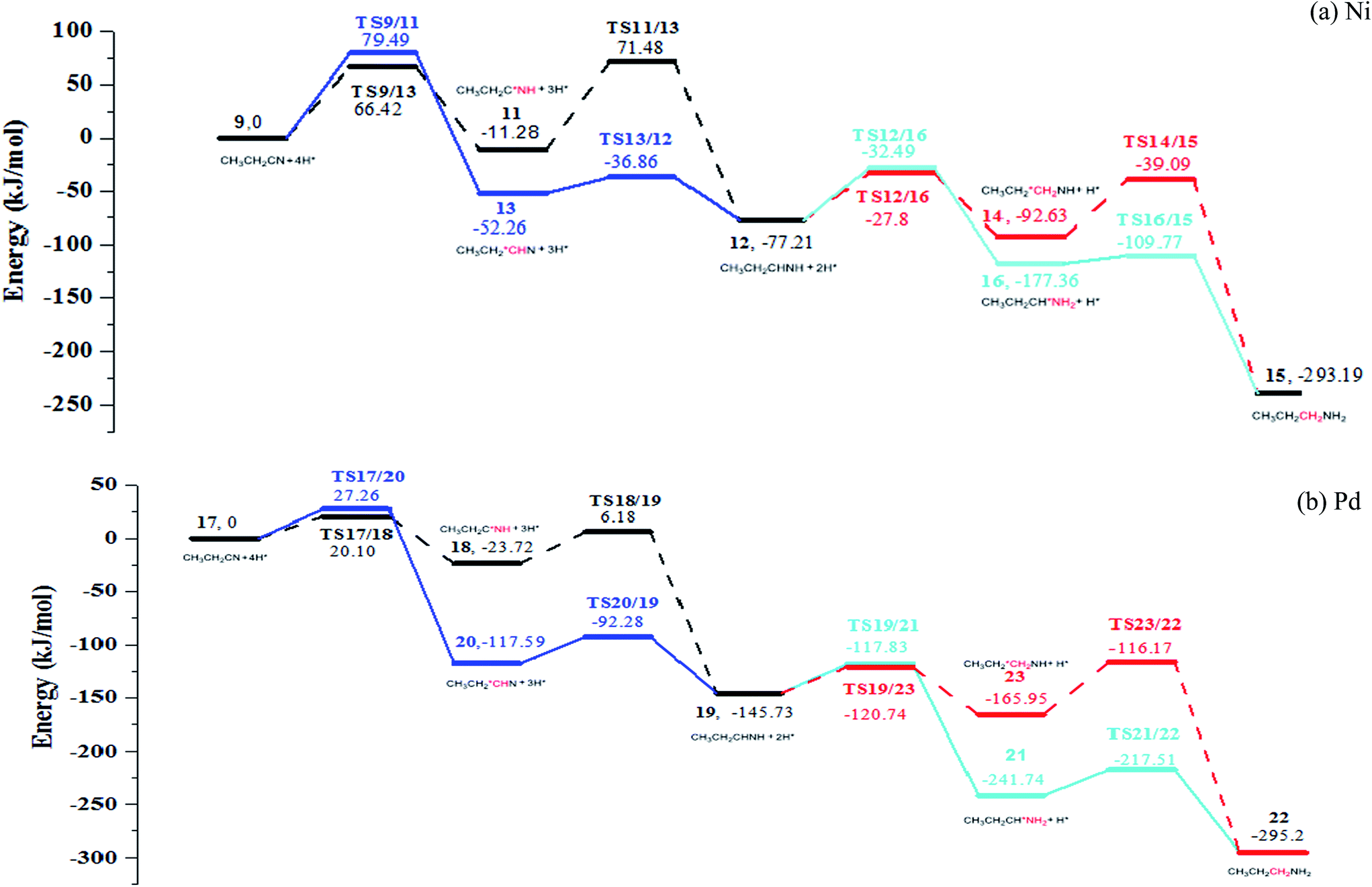
Further hydrogenation of the C3N bond also involves determining of the sequence for formations of the C3–H and N–H bonds. Thus, there are two pathways for both the formation of imine and the further generation of amine: formations of C3–H and N–H in serial or in the reverse sequence, to produce imine; formations of a second C3–H and a second N–H bonds in serial or in the reverse sequence to generate amine. Associated PESs are shown in Fig. 6 (see Fig. S9–S12 in ESI†).
 | ||
| Fig. 6 Potential energy profiles of acrylonitrile further hydrogenation on Ni(111) (a) and Pd(111) (b). | ||
After C3 atom of nitrile group is hydrogenated firstly with the energy barrier of 79.49 kJ mol−1 and 20.10 kJ mol−1 for Ni and Pd respectively, the N atom is attacked by the hydrogen atom with overcoming energy barriers of 82.76 kJ mol−1 and 29.90 kJ mol−1 for Ni(111) and Pd(111) respectively. Upon formation of the C3–H bond, the C3–N bond is rotated, shortening and strengthening the N-metal bonds. Alternatively, if N atom of nitrile group is hydrogenated firstly, energy barriers of 79.49 kJ mol−1 and 27.26 kJ mol−1 are encountered for Ni(111) and Pd(111), respectively. The N–H bond making leads to the rotation of ethyl group, which decreases the steric hindrance of C3 atom. Subsequently, the energy barrier of C3 atom hydrogenation amounts to 15.4 kJ mol−1 and 25.31 kJ mol−1 for Ni(111) and Pd(111) respectively. It is consistent with the expectation. Our calculation exhibits that it is easier for making C3–H bond than N–H bond.
Once obtaining adsorbed imine on metal surface, it undergoes further hydrogenation to generate propylamine. When C3 bonds with a hydrogen atom first, the energy barriers are 44.27 kJ mol−1 and 24.99 kJ mol−1 for Ni(111) and Pd(111), respectively. On the other hand, the energy barriers for forming an N–H bond on Ni(111) and Pd(111) are 53.54 kJ mol−1 and 31.78 kJ mol−1, respectively. Subsequent hydrogenation step corresponds to formation of a N–H bond, with barriers of 49.41 kJ mol−1 and 27.90 kJ mol−1 for Ni(111) and Pd(111), respectively. Finally, for the formation of the C3–H bond, it is energetically much favored (with barriers of 7.59 kJ mol−1 and 24.23 kJ mol−1 for Ni(111) and Pd(111) respectively). It indicated that making a N–H bond is more difficult than to make a C–H bond; most likely it is caused by the interaction of the N lone pair with metal.
The formation of propylene imine on Ni(111) and Pd(111)
As Fig. 2 shown, acrylonitrile may be hydrogenated to get the propylene imine in the proposed pathway. Thus, the formation of propylene imine by the hydrogenation of nitrile group on Ni(111) and Pd(111) are discussed in detail. There are also two pathways for both the formation of propylene imine in Fig. 7 (see Fig. S13–S14 in ESI†): formations of C3–H and N–H in serial (path X) or in the reverse sequence (path Y). Associated PESs are shown in Fig. 7. Compared with potential energy of two pathways, path X is a preferred hydrogenation pathway for Ni and Pd surface. For path X, the C3–H bonds are formed by surmounting barriers of 80.95 and 31.09 kJ mol−1 for Ni and Pd, respectively. Subsequently, the other intermediates TS25/26 and TS28/29 are generated with energy barriers of 29.55 and 91.54 kJ mol−1 for Ni and Pd, respectively. However, no matter whether it is path X or path Y, the energy barriers of formation of propylene imine are much higher than the hydrogenation of acrylonitrile to propionitrile. Therefore, in the hydrogenation of acrylonitrile, the hydrogenation of vinyl group is easier than the hydrogenation of nitrile group, which is a preferred hydrogenation pathway.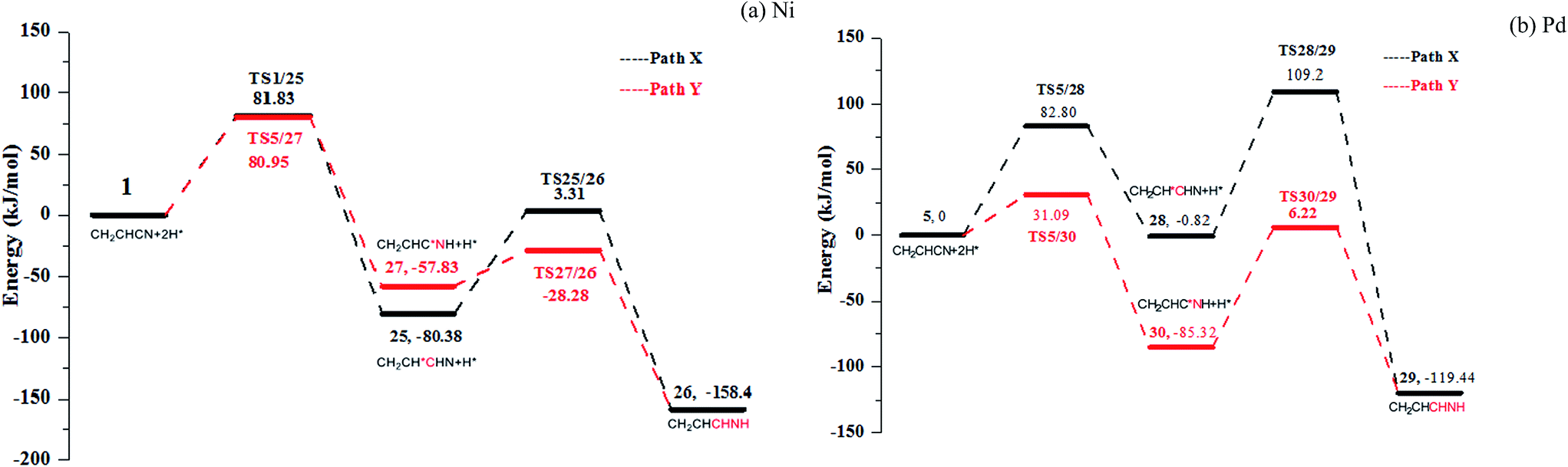 | ||
| Fig. 7 Potential energy profiles of propylene imine formation on Ni(111) (a) and Pd(111) (b). | ||
A comparison of the hydrogenations of acrylonitrile on Ni(111) and Pd(111)
Fig. 8 shows the reaction energy barriers of the hydrogenation of acrylonitrile on Ni(111) and Pd(111). Once the hydrogenation of acrylonitrile is carried out, the energy barriers of the hydrogenation of vinyl is far lower than nitrile. It proves out the hydrogenation of vinyl is preferred pathway for Ni and Pd. Upon comparing different reaction pathways, it is clear that formation of a C2–H bond is always more favorable than making a C1–H bond in the hydrogenation of vinyl group. The result shows that the Ni catalyzed hydrogenation of vinyl is more difficult than Pd. Further, it is obvious that the energy barriers for further hydrogenation on Pd is far lower than on Ni. Thus, the further hydrogenation is more favorable on Pd(111) as compared to that on Ni(111), the selectivity on the former surface should be lower considering the comparable barriers for the two hydrogenation processes.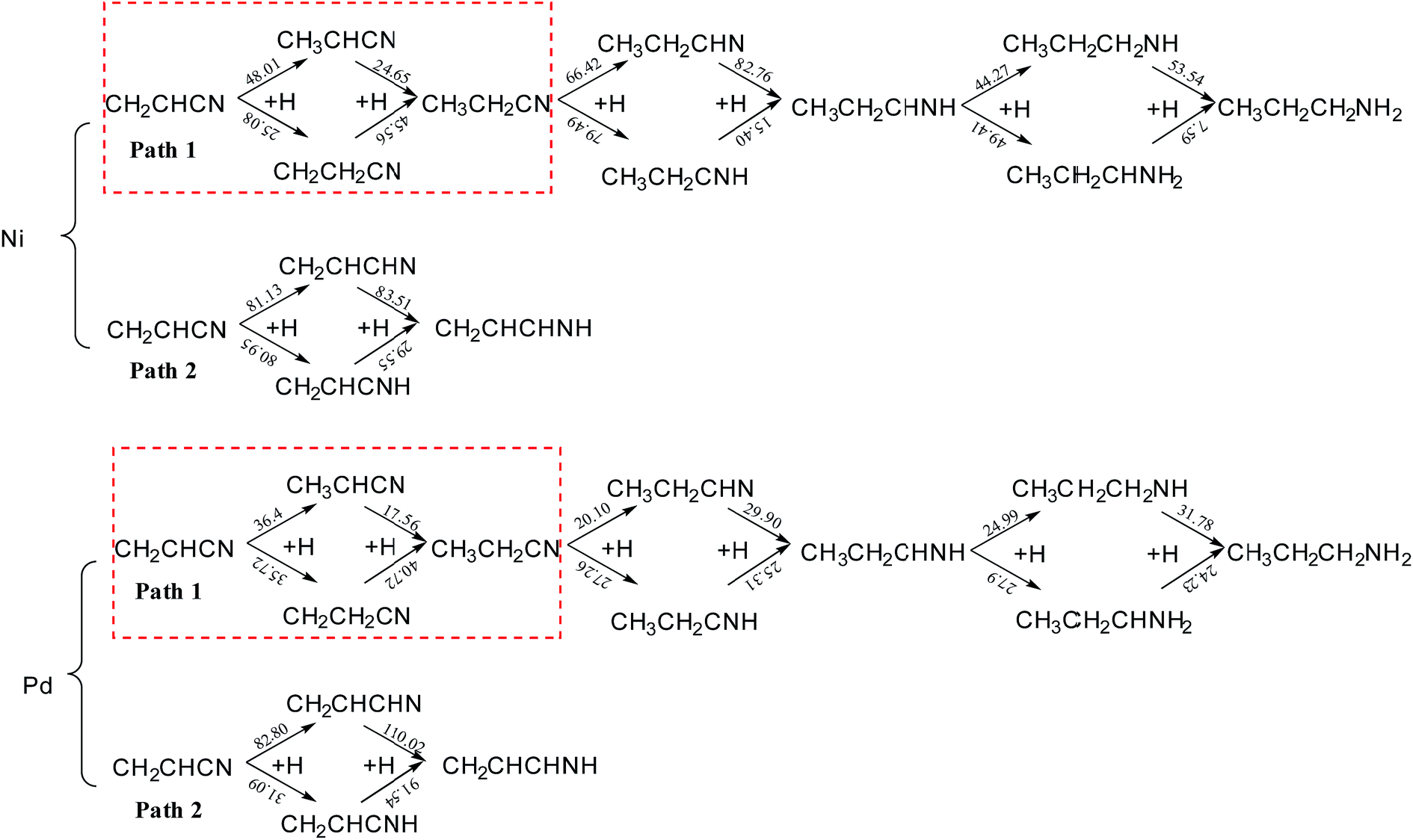 | ||
| Fig. 8 Classical barrier energies for acrylonitrile hydrogenation reactions are shown on the reaction network for Ni(111) and Pd(111). | ||
A comparison of the reaction rates for the hydrogenation of acrylonitrile indicates obviously higher reaction rate catalyzed by Pd/C,15 as shown in Fig. 9, thus in agreement with our theoretical results.
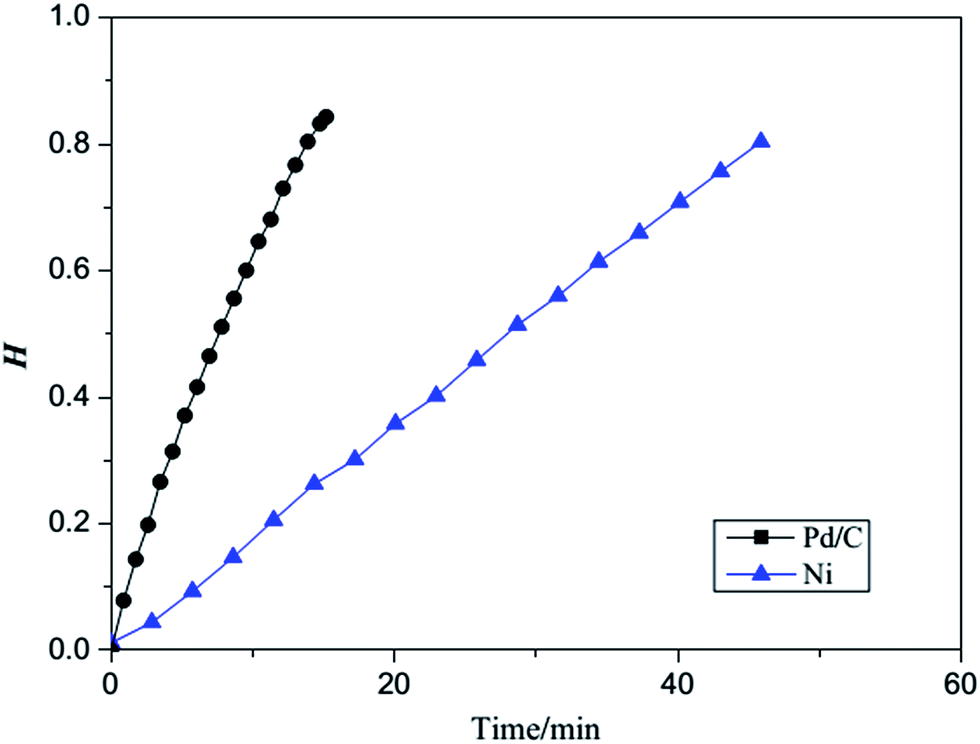 | ||
| Fig. 9 A comparison of the reaction rates for the hydrogenation of acrylonitrile catalyzed RANEY® Ni and Pd/C H = the cumulative hydrogen absorption capacity vs. the theoretical value. | ||
Conclusion
In summary, we have presented the combined experimental and theoretical investigation on the hydrogenation of acrylonitrile catalyzed by RANEY® Ni and Pd/C. The similar most stable adsorption mode the substrate on Ni(111) and Pd(111) surfaces have N-fcc configurations. By comparing different reaction pathways, it turns out that the hydrogenations of both the vinyl and nitrile groups on Pd(111) are more favorable as compared to that on Ni(111). However, the selectivity for producing propionitrile is lower on Pd(111), as further hydrogenation of propionitrile is also favorable. The conclusions obtained from theoretical results is in line with the experimental findings.Acknowledgements
The authors are grateful for the financial support from the Natural Science Foundation of China (21376213, 21476194), the Zhejiang Provincial Public Technology Research of China (2014C31123, 2015C31038), China Postdoctoral Science Foundation funded project (2016M590536) and the Fundamental Research Funds for the Central Universities (JUSRP115A05).References
- P. Pollak, G. Romeder, F. Hagedorn and H. P. Gelbke. “Nitriles” in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- K. Nagaiah, S. J. Kulkarni, M. Subrahmanyam and A. V. R. Rao, Indian J. Chem. Technol., 1994, 1, 356 CAS.
- P. Parent, C. Laffon, G. Tourillon and A. Cassuto, J. Phys. Chem., 1995, 99, 5058 CrossRef CAS.
- (a) Y. Ohtani, A. Yamagishi and M. Fujimoto, Bull. Chem. Soc. Jpn., 1979, 52, 69 CrossRef CAS; (b) H. Henning, M. Dornbach, M. Scheibe, E. Klemm and M. Hunger, Microporous Mesoporous Mater., 2012, 164, 104 CrossRef CAS.
- (a) T. Tihanyi, K. Varga, I. Hannus, I. Kiricsi and P. Fejes, React. Kinet. Catal. Lett., 1981, 18, 449 CrossRef CAS; (b) J. L. Margitfalvi, S. Göbölös, M. Hegedüs and E. Talas, Stud. Surf. Sci. Catal., 1988, 41, 145 CrossRef CAS; (c) J. Choi and N. M. Yoon, Synthesis, 1996, 597 CrossRef CAS; (d) A. F. Barrero, E. J. Alvarez-Manzaneda, R. Chahboun and R. Meneses, Synlett, 1999, 1663 CrossRef CAS; (e) H. Li, H. X. Li and J. F. Deng, Appl. Catal., A, 2000, 193, 9 CrossRef CAS; (f) H. Li, H. X. Li, W. L. Dai and J. F. Deng, Appl. Catal., A, 2001, 207, 151 CrossRef CAS; (g) B. Liu, M. H. Qiao, J. Q. Wang, S. H. Xie, H. X. Li and K. N. Fan, J. Chem. Technol. Biotechnol., 2003, 78, 512 CrossRef CAS.
- P. K. Santra and P. Sagar, J. Mol. Catal. A: Chem., 2003, 197, 37 CrossRef CAS.
- (a) J. Kubota, J. Kondo, K. Domen and C. Hirose, J. Electron Spectrosc. Relat. Phenom., 1993, 64–65, 137 CrossRef CAS; (b) V. M. Geskin, R. Lazzaroni, M. Mertens, R. Jerome and J. L. Bredas, J. Chem. Phys., 1996, 105, 3278 CrossRef CAS; (c) X. Crispin, C. Bureau, V. M. Geskin, R. Lazzaroni, W. R. Salaneck and J. L. Bredas, J. Chem. Phys., 1999, 111, 3237 CrossRef CAS; (d) S. W. Xia, X. A. Xu, H. Yu and H. L. Zhang, Chem. Res. Chin. Univ., 2007, 28, 751 CAS; (e) J. Tornero, H. H. Telle, G. Garcia and A. G. Urena, Phys. Chem. Chem. Phys., 2011, 13, 8475 RSC; (f) S. Diaz-Tendero, M. Alcami and F. Martin, Phys. Chem. Chem. Phys., 2013, 15, 1288 RSC; (g) M. Robledo and S. Diaz-Tendero, J. Phys. Chem. C, 2015, 119, 15125 CrossRef CAS.
- (a) C. Fredriksson, R. Lazzaroni, J. L. Bredas, M. Mertens and R. Jerome, Chem. Phys. Lett., 1996, 258, 356 CrossRef CAS; (b) X. Crispin, R. Lazzaroni, V. Geskin, N. Baute, P. Dubois, R. Jerome and J. L. Bredas, J. Am. Chem. Soc., 1999, 121, 176 CrossRef CAS; (c) C. Bureau, M. Defranceschi, J. Delhalle, G. Deniau, J. Tanguy and G. Lecayon, Surf. Sci., 1994, 311, 349 CrossRef CAS.
- F. de Leon-Perez, R. Miotto and A. C. Ferraz, Braz. J. Phys., 2004, 34, 708 CrossRef CAS.
- Y. Chen and D. G. Vlachos, J. Phys. Chem. C, 2010, 114, 4973 CAS.
- (a) M. Marlo and V. Milman, Phys. Rev. B, 2000, 62, 2899 CrossRef CAS; (b) M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS; (c) G. P. Srivastava and D. Weaire, Adv. Phys., 1987, 36, 463 CrossRef CAS.
- (a) R. A. Vansanten and M. Neurock, Catal. Rev., 1995, 37, 557 CrossRef CAS; (b) B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS; (c) A. Gil, A. Clotet, J. M. Ricart, G. Kresse, M. Garcia-Hernandez, N. Rosch and P. Sautet, Surf. Sci., 2003, 530, 71 CrossRef CAS; (d) M. Gajdos, A. Eichler and J. Hafner, J. Phys.: Condens. Matter, 2004, 16, 1141 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225 CrossRef CAS.
- X. Ge, J. B. Pan, X. Z. Chen, C. Qian and S. D. Zhou, Int. J. Hydrogen Energy, 2016 Search PubMed , Submitted.
- P. A. Sheth, M. Neurock and C. M. Smith, J. Phys. Chem. B, 2003, 107, 2009 CrossRef CAS.
- C. Oliva, C. van den Berg, J. W. Niemantsverdriet and D. Curulla-Ferre, J. Catal., 2007, 245, 436 CrossRef CAS.
- A. Valcarcel, A. Clotet, J. M. Ricart, F. Delbecq and P. Sautet, J. Phys. Chem. B, 2005, 109, 14175 CrossRef CAS PubMed.
- (a) X. Ge, J. B. Pan, C. Qian, L. Feng, Y. B. Chen and X. Z. Chen, Catal. Commun., 2014, 46, 201 CrossRef CAS; (b) X. Ge, C. X. Luo, C. Qian, Z. P. Yu and X. Z. Chen, RSC Adv., 2014, 4, 43195 RSC.
- (a) G. Kresse, Phys. Rev. B, 2000, 62, 8295 CrossRef CAS; (b) G. W. Watson, R. P. K. Wells, D. J. Willock and G. J. Hutchings, J. Phys. Chem. B, 2001, 105, 4889 CrossRef CAS.
- (a) F. Mittendorfer and J. Hafner, J. Phys. Chem. B, 2002, 106, 13299 CrossRef CAS; (b) N. Lopez, Z. Lodziana, F. Illas and M. Salmeron, Phys. Rev. Lett., 2004, 93, 146103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra09295k |
| This journal is © The Royal Society of Chemistry 2016 |