
A pre-lithiated phloroglucinol based 3D porous framework as a single ion conducting electrolyte for lithium ion batteries†
Abstract
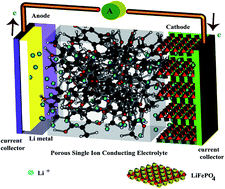
We report the design and synthesis of an inherently porous single ion conducting gel electrolyte made from a pre-lithiated phloroglucinol-terephthalaldehyde 3D framework for lithium ion batteries, adopting a “bottom-up” approach. The cationic transference number of the membrane obtained by blending the complex with PVDF–HFP followed by solution casting was found to be 0.86, close to unity. The mobile lithium ions shuttle through the low resistant pathways offered by the 3D network by virtue of its high porosity. The electrolyte offers a high ionic conductivity of 6.3 × 10−4 S cm−1 at room temperature (22 °C), comparable to the values of most gel polymer electrolytes. Furthermore, the electrolyte membrane displays high thermal stability and good mechanical strength. Coin cells assembled with the membrane perform well at both room temperature and 80 °C.
 Please wait while we load your content...
Please wait while we load your content...

