
Influence of gold nanoparticles applied to catalytic hydrogenation of acetophenone with cationic complexes containing ruthenium†‡
Lanarck C. M. Souzaa,
Thiago A. Santosa,
Cássio R. A. Do Pradoa,
Benedicto A. V. Limab,
Rodrigo S. Corrêa§
b,
Alzir A. Batistab,
Larissa Otuboc,
Javier Ellenad,
Leonardo T. Uenoa,
Luís R. Dinellia and
André L. Bogado*a
aFaculdade de Ciências Integradas do Pontal, Universidade Federal de Uberlândia, Rua vinte, 1600, CEP 38304-402, Ituiutaba, MG, Brazil. E-mail: bogado@ufu.br
bDepartamento de Química, Universidade Federal de São Carlos, CP 676, CEP 13565-905, São Carlos, SP, Brazil
cCentro de Ciência e Tecnologia de Materiais, Instituto de Pesquisas Energéticas e Nucleares, Av. Prof. Lineu Prestes, 2242-Cidade Universitária, CEP: 05508-000, São Paulo, SP, Brazil
dInstituto de Física de São Carlos, Universidade de São Paulo, CP 780, 13560-970, São Carlos, SP, Brazil
First published on 26th May 2016
Abstract
Herein the catalytic activity of cationic ruthenium(II) complexes [Ru]+ is described in the presence of gold nanoparticles (AuNPsn−) in the transfer hydrogenation of acetophenone, to produce phenylethanol. The catalytic activity of the complexes, with a general formula cis-[RuCl(CH3OH)(P–P)(N–N)]+ or cis-[RuCl(CH3OH)(P)2(N–N)]+ {where: P = triphenylphosphine (PPh3); P–P = 1,1-bis(diphenylphosphino)methane (dppm); 1,2-bis(diphenylphosphino)ethane (dppe); 1,3-bis(diphenylphosphino)propane (dppp), 1,4-bis(diphenylphosphino)butane (dppb); N–N = 2,2′-bipyridine; 4,4′-dimethyl-2,2′-bipyridine} was investigated in the presence of AuNPsn−. The interaction between [Ru]+ and AuNPsn− citrate capped is an electrostatic interaction, by a self-assembly processes, to produce a supramolecular species, labeled as [Ru]+/AuNPsn−. This non-covalent interaction has no effect over the chemical and physical chemical parameters of the complexes, which provides a good point of comparison in the presence and absence of AuNPsn−. The AuNPsn− alone have no catalytic activity in the transfer hydrogenation of acetophenone within 24 h of reaction. However, the AuNPsn− have improved the catalytic activity of the complexes that have biphosphines with tensioned or large bite angle, while for the complexes that have biphosphines with a strong chelate effect a decrease in the catalytic activity was observed. The evidence is supported by experimental values of the yields of the hydrogenated product and DFT calculations of the “RuP–P” intermediates. Suitable crystals of cis-[RuCl2(dppe)(bipy)], cis-[RuCl2(dppp)(bipy)] and cis-[RuCl(CH3OH)(dppb)(bipy)](PF6) were obtained and the X-ray structures are presented here.
Introduction
The use of complexes containing ruthenium in the hydrogenation of polar double bonds can occur by two main pathways: by hydrogenation or transfer hydrogenation.1–3 One of the most important mechanisms for H2-hydrogenation of imines and ketones was proposed by R. Noyori4–7 (Nobel Prize laureate in 2001) and some insights were offered by R. H. Morris.6,7 The most used catalyst precursors for this kind of reaction are complexes with phosphine and diamine moieties,4–7 paying special attention to complexes containing ruthenium.8–10Additionally, gold nanoparticles (AuNPsn−), have been used in many areas of knowledge, for example: chemiluminescence sensors,11 cross-link agents,12 colorimetric detection,13 modifier electrodes,14,15 cytotoxicity studies,16 hybrid nanobiomaterials,17–19 biodiagnostics20 and catalysis in liquid phases.17–22 In agreement with the catalytic context, AuNPsn− have also been proposed as a key species in the water–gas-shift reaction (WGSR). This consists of a carboxyl mechanism for the WGSR on the AuNPsn−/CeO2(111) surface, where the pathway for H2 production recombines two H atoms to form H2 on the Au cluster.23–29 Catalytic activity of Au25(SR)18 nanoclusters (R = C2H4Ph) for the aldehyde hydrogenation reaction in the presence of a base has been reported by Kim and co-workers.30 The ability of functionalized AuNPsn− in the reduction of 4-nitrophenol to produce 4-aminophenol is another interesting application of AuNPsn− in catalyses.15,31–39
Recently, we demonstrated that interaction between cationic ruthenium complexes and AuNPsn− does not change the structure and geometry of the complexes, neither the electronic properties of them.12,14 These cationic ruthenium complexes are used as positive charge species in order to aggregate on the surface of negatively charged gold nanoparticles, by a self-assembly processes, which depends only on the concentration of the related species. The maintenance of chemical attributes of the complexes, after interacting with AuNPsn−, provides an application of these new materials in different fields.
Herein the effect of AuNPsn− in the transfer hydrogenation of acetophenone catalyzed by cationic complexes containing ruthenium is described. Suitable crystals of cis-[RuCl2(dppe)(bipy)], cis-[RuCl2(dppp)(bipy)] and cis-[RuCl(CH3OH)(dppb)(bipy)](PF6) were obtained and the X-ray structures are presented here.
Results and discussion
Syntheses and characterization of complexes containing ruthenium
The ruthenium complexes were firstly synthesized as dichloride ruthenium complexes. The designation cis and trans are related to the chloride position in the structure of the complexes. The complexes with a general formula cis-[RuCl2(P–P)(N–N)] {where: P–P = 1,4-bis(diphenylphosphino)butane (dppb), N–N = 2,2′-bipyridine (bipy) or 4,4′-dimethyl-2,2′-bipyridine (Meby)} where synthesized from the binuclear complex containing ruthenium [RuCl2(dppb)]2-μ-(dppb)40,41 as described previously.42–44 The trans-[RuCl2(PPh3)2(bipy)] {where: PPh3 = triphenylphosphine (PPh3)} was prepared from the well-known complex containing ruthenium described by Wilkinson45 [RuCl2(PPh3)3].42,43 The complexes cis-[RuCl2(dppm)(bipy)], cis-[RuCl2(dppe)(bipy)] and cis-[RuCl2(dppp)(bipy)] were synthesized from the trans-[RuCl2(bipy)(PPh3)2] by phosphine exchange. The 31P{1H} NMR spectra of cis-[RuCl2(dppm)(bipy)], cis-[RuCl2(dppe)(bipy)] and cis-[RuCl2(dppp)(bipy)] show two doublets, which are in agreement with the non-magnetic equivalence of the P atoms, suggesting a cis coordination mode of the ligands. The chemical shift of these complexes show an inverse relationship with the Ru–P bond lengths, determined by single crystal X-ray diffraction, where the more shielded phosphorus atoms provide a longer Ru–P bond, and consequently a weaker bond. The anodic peak potential (Epa) observed by cyclic voltammetry (CV) is in agreement with 31P{1H} NMR data and bond lengths for Ru–P. Thus, the higher value of Epa was observed in the CV of the cis-[RuCl2(dppe)(bipy)] (0.73 V), due to the stronger back-donation ruthenium to phosphorus, when compared with the same bond for the cis-[RuCl2(dppp)(bipy)] (see Table 1).| Complex | δa ppm (2JPP = Hz) | Ru–P bond lengthsb (Å) | Epac (V) (RuII/RuIII) |
|---|---|---|---|
| a Using CH2Cl2 as a solvent (D2O capillary), δ with respect to the phosphorus signal of H3PO4 85%.b Obtained by single crystal X-ray diffraction.c CV were carried out at room temperature in CH2Cl2 containing 0.1 mol L−1 Bu4N+ClO4− (TBAP). The working and auxiliary electrodes consisted of stationary Pt foil; the reference electrode was Ag/AgCl in a Luggin capillary. | |||
| Cis-[RuCl2(dppm)(bipy)] | 18.5; 11.2 (64.3) | — | 0.68 |
| Cis-[RuCl2(dppe)(bipy)] | 68.0; 61.0 (54.0) | Ru–P2 (trans Cl) = 2.2465(8) | 0.73 |
| Ru–P1 (trans N2) = 2.2907(9) | |||
| Cis-[RuCl2(dppp)(bipy)] | 40.2; 32.3 (42.1) | Ru–P2 (trans Cl11) = 2.280(3) | 0.69 |
| Ru–P1 (trans N12) = 2.313(3) | |||
The X-ray structures of cis-[RuCl2(dppe)(bipy)] and cis-[RuCl2(dppp)(bipy)] are shown in Fig. 1 and 2, respectively. Selected bond, distances and angles are available in the ESI.† The structures of cis-[RuCl2(dppe)(bipy)] and cis-[RuCl2(dppp)(bipy)] are distorted octahedrally, due to the restricted bite angles of the surrounding bidentate ligands in the molecular structure of the complexes.

The biphosphine bite angle is shorter in the cis-[RuCl2(dppe)(bipy)], when compared with cis-[RuCl2(dppp)(bipy)], P(2)–Ru–P(1) = 84.91(3)° and P(2)–Ru(1)–P(1) = 92.85(10)°, respectively. These results are in agreement with electrochemical and 31P{1H} NMR data. The bond distances and angles47,48 of cis-[RuCl2(dppe)(bipy)] and cis-[RuCl2(dppp)(bipy)] complexes are in the range expected for biphosphine complexes containing ruthenium.42–44
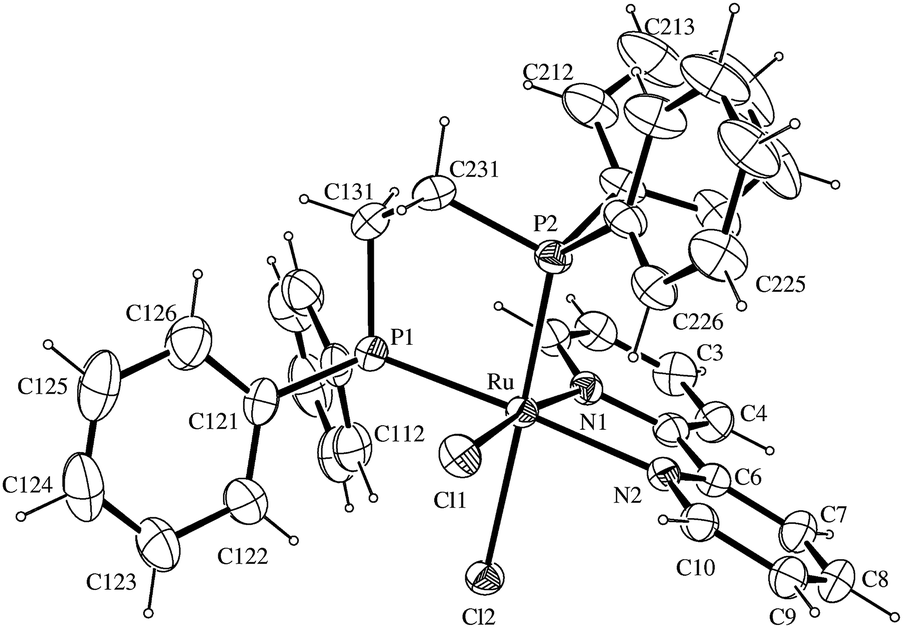
The related cationic ruthenium solvate complexes [RuCl(CH3OH)(dppb)(bipy)]+ (1), [RuCl(CH3OH)(dppb)(Mebipy)]+ (2), [RuCl(CH3OH)(PPh3)2(bipy)]+ (3), [RuCl(CH3OH)(dppm)(bipy)]+ (4), [RuCl(CH3OH)(dppe)(bipy)]+ (5) and [RuCl(CH3OH)(dppp)(bipy)]+ (6), were prepared after adding the dichloro ruthenium complexes (10 μmol each), in methanol (2 mL), within 1 h of magnetic stirring at room temperature. The cis designation in the chemical formulas of cationic complexes is related to the position of coordinated Cl and CH3OH groups (see Scheme 1). Suitable crystals of cis-[RuCl(CH3OH)(dppb)(bipy)](PF6) grown by slow evaporation of diethyl methanol/CH2Cl2 and their structure was determined by single crystal X-ray diffraction (Fig. 3).
 | ||
| Scheme 1 General structures of complexes studied in this work as Cl− salts.50 | ||

The structure of cis-[RuCl(CH3OH)(dppb)(bipy)]+ is also distorted octahedrally, with a methanol molecule coordinated in the trans position of P-atom. Experimental and theoretical study of the kinetics of dissociation in the cis-[RuCl2(dppb)(bipy)] revealed that only the chloride trans to the phosphorus atom of the dppb ligand was dissociated, even in the presence of excess of monodentate ligand, such as monopyridine and functionalized monopyridine.51
Interaction between [Ru]+ and AuNPs−
Recently, we (and other authors) have demonstrated the non-covalent interaction between AuNPsn− and cationic specimens with different applications in many areas of knowledge.12,14,52–54 The process of interaction between cationic ruthenium complexes [Ru]+ and the negative surface of gold nanoparticles (AuNPsn−) can be accompanied by optical measurements and TEM images. The plasmon band adsorption of AuNPsn− occurs at 520 nm, and the addition of [Ru]+ specimens produce an enlarged band, centralized at 625 nm, with a significant decrease in the original plasmon band. The polarization of the conduction electron oscillations in adjacent AuNPsn− causes a red-shift on the plasmon absorbance, attributed to the coupling of plasmon absorbance of the particles.55,56 The time dependence of the interaction between [Ru]+ and AuNPsn− was carried out using optical measurements (UV/vis), with temperature control, using the cationic complex containing ruthenium with formula [RuCl(py)(dppb)(bipy)]+ {where py = pyridine}.12,14 Stock solutions of the cationic complexes were prepared in acetone (5.3 × 10−5 mol L−1) and 100 μL of that was added to a colloidal suspension of AuNPsn− (3 mL, 0.05 mol L−1). The kinetics of interaction between AuNPsn− with [Ru]+ was investigated by monitoring the changes in the electronic spectra as a function of time and temperature in the range from 20 °C to 35 °C (Fig. 4). | ||
| Fig. 4 Time dependence of coupled plasmon band of AuNPsn− (3 mL; 0.05 mol L−1) in the presence of [RuCl(py)(dppb)(bipy)]+ (100 μL, 5.3 × 10−5 mol L−1) as a function of time λ = 625 nm; temperature: 20, 25 and 30 °C. The label inside 20 °C UV/vis: a = induction time, b = flocculation (t = 25 min), c = aggregation. | ||
Herein the terms used by Whitesides and co-workers,57 are adopted. They use flocculation to refer to the instability of colloidal dispersions, agglomeration for the cases involving reversible association of nanoparticles and aggregation for irreversible association.
In Fig. 4, it can be observed that the induction and flocculation period (a and b) is dependent on the temperature range, and at 30 °C the flocculation period practically disappears, and the interaction between AuNPsn− and [Ru]+ species go directly to the aggregation. Bellino and co-workers58 investigated the kinetics of interaction of citrate stabilized gold nanoparticles with negatively and positively charged mercapto ligands. Another interesting aspect is the possibility of controlling the stabilization/destabilization of AuNPsn− with pentacyanoferrate(II) ions.56 In both cases, the kinetics proceed relatively fast, leading to the decay of the 520 nm band and the rise of the plasmon coupling band at 650 nm, a characteristic of flocculation. In a typical experiment using the Arrhenius equation in its linear form, the logarithmic of kinetic constants, obtained by the initial rate method, were plotted against the inverse of temperature (Fig. 5).
 | ||
| Fig. 5 Arrhenius plot. (A) Flocculation period. (B) Aggregation period. | ||
The activation energy (Ea) and the pre-exponential factor (A) were obtained related to flocculation and aggregation period (Table 2). The Ea of the flocculation period is 46.14 kJ mol−1, which is higher than the Ea of the aggregation period, 38.88 kJ mol−1. It is reasonable that the Ea to promote a flocculation should be higher than the Ea to promote an aggregation, since the species should meet each other, with appropriate kinetic energy to produce an aggregate. The pre-exponential factor is also in conformity, the A value of the flocculation period is almost one thousand times higher than the aggregation period.
| Coupled plasmon band at λ = 625 nm | Ea (kJ mol−1) | ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A A |
A |
|---|---|---|---|
| Flocculation period | 46.14 | 13.157 | 517621 |
| Aggregation period | 38.88 | 6.32 | 556 |
TEM images of the flocculation period were obtained, where the [RuCl(py)(dppb)(bipy)]+ (0–1000 μL, 5.3 × 10−5 mol L−1 in acetone) was added to a colloidal suspension of AuNPsn− (1.0 mL, 0.05 mol L−1) (Fig. 6). The TEM images show an average diameter of the AuNPsn− of approximately 12 nm and reveal that they did not grow in a radial mode when the [Ru]+ species are present, but in the flocculation period, the approximation of the spherical nanoparticles is observed. It is in agreement with the optical measurements observed by UV/vis, with the appearance of the coupled plasmon band at 625 nm. A precipitate is formed in the presence of a large excess of the [Ru]+, labeled as an assembly of AuNPsn− and the [Ru]+ ([Ru]+/AuNPsn−), and it is observed as round-shaped bright spots, which is soluble in 2-propanol, as previously published.12,14
 | ||
| Fig. 6 TEM images of the flocculation period between a colloidal suspension of AuNPsn− (0.05 mol L−1) and [RuCl(py)(dppb)(bipy)]+ (0–1000 μL), 5.3 × 10−5 mol L−1 in acetone. | ||
Catalytic activity of the complexes and DFT calculation
It can be observed in the Table 3 that AuNPsn− has no catalytic activity in the hydrogenation of acetophenone, with the applied conditions, within 5.5 h of reaction. However, it can improve the catalytic activity of some biphosphine complexes containing ruthenium, within 1 h of reaction. When the (1)/AuNPsn− was used as a pre-catalyst, it produced 24% more phenylethanol than (1) alone. The time dependence of (1)/AuNPsn− showed that the quantitative amount of product can be obtained after 2 h of reaction, and the best molar relationship between (1) and AuNPsn− was 2.0/1.0 respectively, as described in the caption in Table 3. When the (2)/AuNPsn− was used as a pre-catalyst, it produced almost 12% more product. There is no significant improvement when (3) was used as a pre-catalyst. In this case, in the presence or absence of AuNPsn−, the amount of formed product is almost the same, with an average of 81.69% of product. When the (3)/AuNPsn− was used as a pre-catalyst, with an additional amount of PPh3 (5 eq. in respect of the ruthenium complex), a drastic decrease in the catalytic activity was observed, only 3% of the product was observed after 24 h of reaction. In the first moment, it suggests that an intermediate species with an uncoordinated phosphine, or a dangling biphosphine, could be the real catalyst of that reaction, and the AuNPsn− could be acting as a co-catalyst in the transfer hydrogen pathway from isopropanol to substrate.
|
||||
|---|---|---|---|---|
| Pre-catalyst | Phenylethanol% | TOF (h−1) | ||
| Without AuNPsn− | With AuNPsn− | Without AuNPsn− | With AuNPsn− | |
| a Applied ruthenium complexes [RuCl(CH3OH)(dppb)(bipy)]+ (1); [RuCl(CH3OH)(dppb)(Mebipy)]+ (2); [RuCl(CH3OH)(PPh3)2(bipy)]+ (3); [RuCl(CH3OH)(dppm)(bipy)]+ (4); [RuCl(CH3OH)(dppe)(bipy)]+ (5); [RuCl(CH3OH)(dppp)(bipy)]+ (6). Every reaction was carried out in threefold analysis. TOF = turnover frequency = mol phenylethanol/mol [Ru]+/h. Time = 1 h, except in (a) t = 5.5 h, and (b) t = 24 h. AuNPsn− = 10 mL (0.05 mol L−1). Molar relationship [Ru]+/AuNPsn−/acetophenone/KOH = 1/0.5/1000/20. | ||||
| AuNPsn−(a) | — | 4.01 | — | — |
| (1) | 48.75 | 77.74 | 487 | 777 |
| (2) | 72.95 | 84.40 | 729 | 875 |
| (3) | 87.48 | 75.90 | 875 | 759 |
| (4) | 24.10 | 71.23 | 240 | 710 |
| (5) | 93.54 | 24.20 | 930 | 240 |
| (6) | 50.53 | 35.94 | 500 | 350 |
| (3) + 5 eq. PPh3(b) | — | 3.00 | — | — |

To better understand this behavior, the ΔG° energy to produce a dangling biphosphine was determined by DFT calculation (see Scheme 2). The values of ΔG° are described in the Scheme 2, with positive values of ΔG° in all cases, which suggest that dissociation of one side of P–P is a non-spontaneous process. On the other hand, the coordination κ2-P–P mode is a spontaneous process.
 | ||
| Scheme 2 Thermodynamic stability of the complexes [RuCl(CH3OH)(dppb)(bipy)]+ (1), [RuCl(CH3OH)(dppm)(bipy)]+ (4), [RuCl(CH3OH)(dppe)(bipy)]+ (5) and [RuCl(CH3OH)(dppp)(bipy)]+ (6) due to the dissociation of one side of P–P. | ||
In Fig. 7, it can be observed that the catalytic activity tendency, without AuNPsn−, follows the thermodynamic stability of the applied complexes containing ruthenium, due to the structure of a dangling biphosphine. However, the tendency is opposite when AuNPsn− is applied to the catalytic system. The catalytic activity of the precursors (5) and (6) decrease in the presence of AuNPsn−, which has support in the thermodynamic stability of the complexes. As they have a higher stability, the interaction with AuNPsn− should be more effective, therefore the catalytic performance decreases. For the sake of illustration, the precursor (5) exhibits the higher ΔG° value among the studied precursors, with 108.3 kJ mol−1 due to the η1-P–P coordination mode. This suggests that (5) is a stable complex, as expected by the coordination chemistry knowledge, and it showed the best catalytic activity without AuNPsn−, with 93% of the phenylethanol after 1 h of reaction.
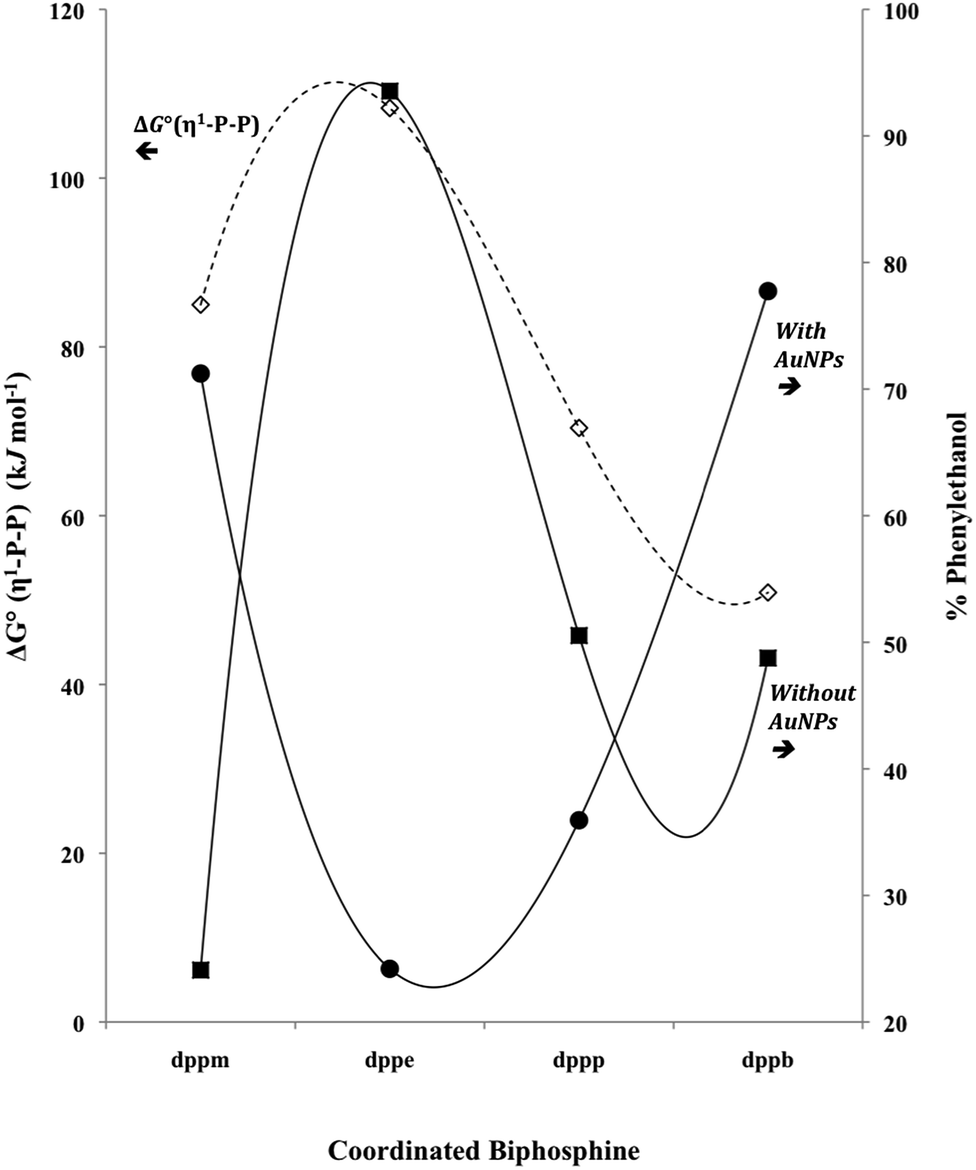 | ||
| Fig. 7 Relationship between standard free-Gibbs energies for one site dissociation of P–P ligand (dashed line) and amount of catalytic product (solid lines: ■ = without AuNPsn−, ● = with AuNPsn−). | ||
More outstanding results were obtained from precursors (1) and (4) in the presence of AuNPsn−, suggesting that these nanoparticles can improve the catalytic activity of the complexes that have biphosphines with tensioned or large bite angle. In some way, the AuNPsn− can stabilize the structure of these complexes, strongly suggested by η1-P–P type coordination. Thus, these reaction intermediates can act as effective catalyst precursors. Therefore, there seem to be two different pathways acting in the catalytic systems of those complexes: one related to dppe and dppp, and the other to dppm and dppb complexes containing ruthenium.
The Cl− dissociation was also investigated by DFT in the complexes with general formula [RuCl(CH3OH)(κ2-P–P)(bipy)]+, and the energy to produce the dicationic complexes was surprisingly over 800 kJ mol−1. It suggests that the real catalyst should keep at least one anionic ligand coordinated in the metal centre, which could be the Cl−, or a hydride species formed in situ.
The dependence of the product on the P–P ligand was also observed in the reactions of [RuCl3(NO)(P–P)] complexes with 2-mercaptopyridine (pyS) ligand.59 When the P–P ligand was dppen {1,2-bis(diphenylphosphino)ethylene}, dppe or dppp, the only product was the corresponding [Ru(pyS)2(P–P)] complexes. However, with dppm and dppb a different pattern of reactivity was found and the main product was identified as [Ru(pyS)2(NO)(η1-P–PO)]PF6.
Experimental
Reagents
All reactions were carried out under an argon atmosphere using standard Schlenk techniques. RuCl3·xH2O, H[AuCl4], triphenylphosphine (PPh3), 1,1-bis(diphenylphosphino)methane (dppm), 1,2-bis(diphenylphosphino)ethane (dppe), 1,3-bis(diphenylphosphino)propane (dppp), 1,4-bis(diphenylphosphino)butane (dppb), 2,2′-bipyridine (bipy), 4,4′-dimethyl-2,2′-bipyridine (Mebipy), pyridine (py), 4′-methylpyridine (Mepy), 4′-tert-butylpyridine (tbut-py), 4′-vinylpyridine and sodium citrate were purchased from Aldrich and used as received. Reagent grade solvents were distilled prior to use.Instrumentation
Ruthenium complexes were analyzed by 31P{1H} NMR on an ARX 200 MHz and a DRX 400 MHz Bruker instrument. Samples were prepared under an inert atmosphere of argon and analyzed at room temperature with a D2O capillary and dichloromethane (CH2Cl2) as solvent. Chemical shifts are relative to the signal of H3PO4, 85%, as an external reference. Cyclic voltammetry experiments were carried out at 25 °C in CH2Cl2 containing 0.1 mol L−1 Bu4N+ClO4− (TBAP), with a Bioanalytical System Inc. BAS-100B/W electrochemical analyzer. The working and auxiliary electrodes were stationary Pt foil; a Luggin capillary probe was used and the reference electrode was Ag/AgCl. Under the conditions used, E0 for the one-electron oxidation of [Fe(η5-C5H5)2], added to the test solutions as an internal reference, is +0.43 V. Elemental analyses were performed at the Department of Chemistry at the Federal University of São Carlos, (Brazil), with a FISIONS CHNS EA1108 micro analyzer. Suitable crystals for X-ray analyses were grown by slow evaporation of a dichloromethane-diethyl ether solution of cis-[RuCl2(dppe)(bipy)], cis-[RuCl2(dppp)(bipy)] and cis-[RuCl(CH3OH)(dppb)(bipy)](PF6).The syntheses of gold nanoparticles and the aggregation with cationic ruthenium complexes were controlled by UV/vis analyses on a Shimadzu UV spectrophotometer model UV-1800 with a Shimadzu temperature controlled cell hold model TCC-100.
Transmission electron microscopy (TEM) images were obtained using a JEOL JEM 2100 operating at 200 kV. The sample was prepared as follows: stock solutions of the cationic complexes were prepared in acetone (5.3 × 10−5 mol L−1) and 100 μL of this was added to a colloidal suspension of AuNPsn− (3 mL, 0.05 mol L−1). The resulting solution was dropped onto a 400 mesh copper grid coated with collodion film.
All calculations were carried out using the Gaussian09 suite of programs.60 The structures were optimized by the Density Functional Theory (DFT) method, using the B3LYP hybrid functional, which includes the non-local exchange term with three parameters of Becke and the correlation term of Lee–Yang–Parr.61,62 The basis set used to build the molecular orbitals were the Los Alamos effective core potential (ECP) and double-zeta valence basis set (LanL2DZ) for ruthenium and 6-311+G(d,p) for the remaining atoms. The Hessian matrix was calculated for the optimized structures in order to verify the nature of the stationary state.
X-ray diffraction data
The crystals were mounted on an Enraf-Nonius Kappa-CCD diffractometer with graphite monochromated MoKα (λ = 0.71073 Å) radiation. The final unit cell parameters were based on all reflections. Data collections for all complexes were carried out at room temperature (293 K), with the COLLECT program;63 integration and scaling of the reflections were performed with the HKL Denzo–Scalepack system of programs.64 The crystal structures were solved by the direct method using SHELXS-97 (ref. 65) and refined anisotropically (non-hydrogen atoms) by full-matrix least-squares on F2 using a SHELXL-97 (ref. 65) program. A Gaussian method implemented was used for the absorption corrections.66 All hydrogen atoms were positioned stereochemically and refined using the riding model. The program ORTEP-3 (ref. 46) was used to draw the molecules. Table 1, in the ESI,† summarizes the data collection and experimental details of cis-[RuCl2(dppe)(bipy)], cis-[RuCl2(dppp)(bipy)] and cis-[RuCl(CH3OH)(dppb)(bipy)](PF6).Preparation of gold nanoparticles (AuNPsn−)
AuNPsn− with a diameter of 10–18 nm were prepared by citrate reduction of H[AuCl]4 in aqueous solution according to a well-known method described by Frens.67 To summarize, 20 μL of solution containing H[AuCl]4 (Au 58%) was added to 100 mL of water. The resulting solution was brought to reflux, and 3 mL of sodium citrate solution (1%) was introduced while stirring. The solution was then kept boiling for another 30 min, while the colors changed from yellow to deep blue to red. Finally, the solution was left to cool to room temperature.Synthesis of ruthenium complexes
Kinetic measurements
The kinetics of interaction between AuNPsn− and [Ru]+ was investigated by monitoring the changes in the electronic spectra as a function of time and temperature in the range from 20 °C to 35 °C. In a typical experiment using [RuCl(py)(dppb)(bipy)]+, the complex is described as follow: a stock solution of the cationic complex was prepared in acetone (5.3 × 10−5 mol L−1) and 100 μL of that was added to a colloidal suspension of AuNPsn− (3 mL, 0.05 mol L−1). The time dependence of coupled plasmon band of AuNPsn− was recorded at λ = 625 nm by UV/vis analyses on a Shimadzu UV spectrophotometer model UV-1800 with a Shimadzu temperature controlled cell hold model TCC-100.[Ru]+/AuNPsn− preparation and catalyses
The dichloride ruthenium complexes were stirred for 1 hour in methanol, and after that, a colloidal suspension of AuNPsn− (10 mL, 0.05 mol L−1) was added, and the resulting mixture was magnetically stirred for 10 min. The solvent was evaporated until dry, under vacuum, and the obtained solid, labeled as [Ru]+/AuNPsn−, was characterized as a supramolecular species, as described previously.12,14 The [Ru]+/AuNPsn− were applied to the transfer hydrogenation reaction of acetophenone, and a typical catalytic experiment is described as follows: the [Ru]+/AuNPsn− was dissolved in 2-propanol (10 mL), and acetophenone (10 mmol, 0.2 mol L−1) was added, in the presence of KOH (0.2 mol L−1 in 2-propanol). The catalytic reactions were carried out in argon atmosphere at 82 °C. Molar relationship [Ru]+/AuNPsn−/acetophenone/KOH = 1/0.5/1000/20. The reaction products were analyzed by gas chromatography using a Thermo Scientific GC – Focus chromatograph equipped with a FID detector. A LM-120 column (poly(ethyleneglycol), 25 m long, 0.25 mm i.d. 0.25 μm film thickness) was used to characterize acetophenone and phenylethanol. Hexadecane was used as an internal standard and N2 was the gas carrier (2.0 mL min−1). The temperature program was from 170 °C (2 min) to 200 °C (2 min) at a heating rate of 10 °C min−1.Conclusions
The interaction between [Ru]+ and AuNPsn− citrate capped is an electrostatic interaction, by self-assembly processes, to produce a supramolecular species, labeled as [Ru]+/AuNPs−. The time dependence of the interaction between [Ru]+ and AuNPsn− was investigated by optical measurements (UV/vis), with three different steps in solution: induction, flocculation and aggregation periods. All of these three steps are dependent on the temperature range; at 30 °C the flocculation period practically disappeared, and the interaction between AuNPsn− and [Ru]+ specimens go directly to the aggregation. The Arrhenius parameters of the coupled plasmon band at λ = 625 nm reveal that the flocculation period is energetically higher than the aggregation period, and TEM images reveal that the AuNPsn− did not grow in a radial mode when the [Ru]+ species were present. It provides only the approximation of the spherical nanoparticles, which are in agreement with the coupled plasmon band in the flocculation period observed by UV/vis. Therefore, the kinetic energy acquired in the flocculation period promotes the meeting of [Ru]+ and AuNPsn−, and it has enough energy to promote the aggregation of the specimens in solution, with a decrease in the A value.This non-covalent interaction has no effect over the chemical and physical chemical parameters of the complexes, which provide a good point of comparison in the presence and absence of AuNPsn−, when these species are applied to catalysis. The AuNPsn− alone has no catalytic activity in the transfer hydrogenation of acetophenone within 24 h of reaction. However, the AuNPsn− improved the catalytic activity of the complexes that have biphosphines with a tensioned or large bite angle, while the complexes that have biphosphines with a strong chelate effect, a decrease in the catalytic activity was observed. The evidence is supported by experimental values of the yields of the hydrogenated product and DFT calculation of the “RuP–P” intermediates. In general, complexes with lower values of  are less stable in their chelate mode, and therefore these complexes showed a decrease in the catalytic activity, however in the presence of AuNPsn−, the inverse behavior was observed.
are less stable in their chelate mode, and therefore these complexes showed a decrease in the catalytic activity, however in the presence of AuNPsn−, the inverse behavior was observed.
Acknowledgements
We would like to thank FAPEMIG, CNPq, CAPES, FAPESP, FINEP, DAAD, and RQ-MG for their financial support. The authors are also thankful to the Grupo de Materiais Inorgânicos do Triângulo (GMIT) research group supported by FAPEMIG.Notes and references
- C. Elschenbroich, Organometallics, Wiley-VCH, Weinheim, 3rd edn, 2006 Search PubMed.
- D. Steinborn, Fundamentals of Organometallic Catalysis, Wiley-VCH, Weinheim, 1st edn, 2012 Search PubMed.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, New Jersey, 4th edn, 2005 Search PubMed.
- R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345 CrossRef CAS.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS.
- K. Abdur-Rashid, A. J. Lough and R. H. Morris, Organometallics, 2001, 20, 1047 CrossRef CAS.
- K. Abdur-Rashid, S. E. Clapham, A. Hadzovic, J. N. Harvey, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2002, 50, 15104 CrossRef.
- M. P. de Araujo, A. T. de Figueiredo, A. L. Bogado, G. von Poelhsitz, J. Ellena, E. E. Castellano, C. L. Donnici, J. V. Comasseto and A. A. Batista, Organometallics, 2005, 24, 6159 CrossRef CAS.
- T. F. Gallati, A. L. Bogado, G. von Poelhsitz, J. Ellena, E. E. Castellano, A. A. Batista and M. P. de Araujo, J. Organomet. Chem., 2007, 692, 5447 CrossRef.
- D. A. Cavarzan, F. D. Fagundes, O. Fuganti, C. W. P. da Silva, C. B. Pinheiro, D. F. Back, A. Barison, A. L. Bogado and M. P. de Araujo, Polyhedron, 2013, 62, 75 CrossRef CAS.
- E. Rampazzo, S. Bonacchi, D. Genovese, R. Juris, M. Marcaccio, M. Montalti, F. Paolucci, M. Sgarzi, G. Valenti, N. Zaccheroni and L. Prodi, Coord. Chem. Rev., 2012, 256, 1664 CrossRef CAS.
- V. F. Ferreira, C. R. A. do Prado, C. M. Rodrigues, L. Otubo, A. A. Batista, J. W. da Cruz Jr, J. Ellena, L. R. Dinelli and A. L. Bogado, Polyhedron, 2014, 78, 46 CrossRef CAS.
- R. Manjumeena, D. Duraibabu, T. Rajamuthuramalingam, R. Venkatesan and T. Kalaichelvan, RSC Adv., 2015, 5, 69124 RSC.
- K. M. de Oliveira, T. C. C. dos Santos, L. R. Dinelli, J. Z. Marinho, R. C. Lima and A. L. Bogado, Polyhedron, 2013, 50, 410 CrossRef CAS.
- A. Ahmad, Y. wei, F. Syed, M. Imran, Z. U. H. Khan, K. Tahir, A. U. Khan, M. Raza, Q. Khan and Q. Yuan, RSC Adv., 2015, 5, 99364 RSC.
- F. J. Osonga, I. Yazgan, V. Kariuki, D. Luther, A. Jimenez, P. Le and O. A. Sadik, RSC Adv., 2016, 6, 2302 RSC.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2001, 40, 4128 CrossRef CAS.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042 CrossRef CAS PubMed.
- N. L. Rosi and C. A. Mirkin, Nanostructures in Biodiagnostics, Chem. Rev., 2005, 105, 1547 CrossRef CAS PubMed.
- N. Yan, C. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179 CrossRef CAS.
- L. Liu, Y. Zhao, Q. Chen, X. Shi and M. Shen, RSC Adv., 2015, 5, 104239 RSC.
- Y. Chen, H. Wang, R. Burch, C. Hardacre and P. Hu, Faraday Discuss., 2011, 152, 121 RSC.
- L. C. Grabow, A. A. Gokhale, S. T. Evans, J. A. Dumesic and M. Mavrikakis, J. Phys. Chem. C, 2008, 112, 4608 CAS.
- A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, J. Am. Chem. Soc., 2008, 130, 1402 CrossRef CAS PubMed.
- Z. P. Liu, S. J. Jenkins and D. A. King, Phys. Rev. Lett., 2005, 94, 196102 CrossRef PubMed.
- J. A. Rodriguez, P. Liu, J. Hrbek, J. Evans and M. Perez, Angew. Chem., Int. Ed., 2007, 46, 1329 CrossRef CAS PubMed.
- X. M. Liu, Z. M. Ni, P. Yao, Q. Xu, J. H. Mao and Q. Q. Wang, Acta Phys.–Chim. Sin., 2010, 26, 1599 CAS.
- L. Barrio, P. Liu, J. A. Rodriguez, J. M. Campos-Martin and J. L. G. Fierro, J. Chem. Phys., 2006, 125, 164715 CrossRef CAS PubMed.
- G. Li, H. Adroshan, Y. Chen, R. Jin and H. J. Kim, J. Am. Chem. Soc., 2015, 137, 14295 CrossRef CAS PubMed.
- N. Li, P. Zhao, M. E. Igartua, A. Rapakousiou, L. Salmon, S. Moya, J. Ruiz and D. Astruc, Inorg. Chem., 2014, 53, 11802 CrossRef CAS PubMed.
- N. Pradhan, A. Pal and T. Pal, Colloids Surf., A, 2002, 196, 247 CrossRef CAS.
- A. Shivhare, S. J. Ambrose, H. Zhang, R. W. Purves and R. W. J. Scott, Chem. Commun., 2013, 49, 276 RSC.
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577 RSC.
- P. Pachfule, S. Kandambeth, D. D. Díaz and R. Banerjee, Chem. Commun., 2014, 50, 3169 RSC.
- Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467 CrossRef CAS PubMed.
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908 CrossRef CAS.
- N. Pradhan, A. Pal and T. Pal, Langmuir, 2001, 17, 1800 CrossRef CAS.
- K. Esumi, R. Isono and T. Yoshimura, Langmuir, 2004, 20, 237 CrossRef CAS PubMed.
- M. Bressan and P. Rigo, Inorg. Chem., 1975, 14, 2286 CrossRef CAS.
- C. W. Jung, P. E. Garrou, P. R. Hoffman and K. G. Caulton, Inorg. Chem., 1984, 23, 726 CrossRef CAS.
- A. A. Batista, M. O. Santiago, C. L. Donnici, I. S. Moreira, P. C. Healy, S. L. Berners-Price and S. L. Queiroz, Polyhedron, 2001, 20, 2123 CrossRef CAS.
- A. A. Batista, E. A. Polato, S. L. Queiroz, O. R. Nascimento, B. R. James and S. J. Rettig, Inorg. Chim. Acta, 1995, 230, 111 CrossRef CAS.
- S. L. Queiroz, A. A. Batista, G. Oliva, M. T. do P. Gambardella, R. H. A. Santos, K. S. MacFarlane, S. J. Rettig and B. R. James, Inorg. Chim. Acta, 1998, 267, 209 CrossRef CAS.
- T. A. Stepheson and G. Wilkinson, J. Inorg. Nucl. Chem., 1966, 28, 945 CrossRef.
- L. J. Farrugia, ORTEP3 for Windows, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- Selected bond lengths and angles of cis-[RuCl2(dppe)(bipy)] showing the atom labeling and 50% probability ellipsoids. Bond lengths (Å): Ru–N(1) 2.070(3), Ru–N(2) 2.129(3), Ru–P(2) 2.2465(8), Ru–P(1) 2.2907(9), Ru–Cl(1) 2.4267(9), Ru–Cl(2) 2.4815(8). Bond angles (°): N(1)–Ru–N(2) 77.75(10), N(1)–Ru–P(2) 92.67(7), N(2)–Ru–P(2) 98.99(7), N(1)–Ru–P(1) 99.21(8), N(2)–Ru–P(1) 175.11(7), P(2)–Ru–P(1) 84.91(3), N(1)–Ru–Cl(1) 172.10(8), N(2)–Ru–Cl(1) 94.42(8), P(2)–Ru–Cl(1) 89.62(3), P(1)–Ru–Cl(1) 88.52(4), N(1)–Ru–Cl(2) 88.91(7), N(2)–Ru–Cl(2) 81.90(7), P(2)–Ru–Cl(2) 178.32(3), P(1)–Ru–Cl(2) 94.27(3), Cl(1)–Ru–Cl(2) 88.90(3).
- Selected bond lengths and angles of cis-[RuCl2(dppp)(bipy)] showing the atom labelling and 30% probability ellipsoids. Bond lengths (Å): Ru(1)–N(11) 2.100(8), Ru(1)–N(12) 2.117(9), Ru(1)–P(2) 2.280(3), Ru(1)–P(1) 2.313(3), Ru(1)–Cl(11) 2.426(3), Ru(1)–Cl(12) 2.476(3). Bond angles (°): N(11)–Ru(1)–N(12) 77.3(4), N(11)–Ru(1)–P(2) 97.9(2), N(12)–Ru(1)–P(2) 92.0(2), N(11)–Ru(1)–P(1) 105.0(3), N(12)–Ru(1)–P(1) 174.3(2), P(2)–Ru(1)–P(1) 92.85(10), N(11)–Ru(1)–Cl(11) 170.2(3), N(12)–Ru(1)–Cl(11) 93.2(3), P(2)–Ru(1)–Cl(11) 84.70(10), P(1)–Ru(1)–Cl(11) 84.29(10), N(11)–Ru(1)–Cl(12) 84.1(2), N(12)–Ru(1)–Cl(12) 82.6(2), P(2)–Ru(1)–Cl(12) 173.79(11), P(1)–Ru(1)–Cl(12) 92.33(10), Cl(11)–Ru(1)–Cl(12) 92.40(9). The data of the left structure see ESI.†.
- Selected bond lengths and angles of [RuCl(dppb)(bipy)(CH3OH)]+ showing the atom labeling and 30% probability ellipsoids. Bond lengths (Å): Ru(1)–N(1) 2.083(3), Ru(1)–N(2) 2.134(3), Ru(1)–P(1) 2.2816(11), Ru(1)–P(2) 2.3381(10), Ru(1)–Cl(1) 2.4198(10), Ru(1)–O(1s) 2.249(3), O(1s)–C(1s) 1.437(6). Bond angles (°): N(1)–Ru(1)–N(2) 78.22(12), N(1)–Ru(1)–O(1s) 82.11(11), N(2)–Ru(1)–O(1s) 81.31(11), N(1)–Ru(1)–P(1)105.60(9), N(2)–Ru(1)–P(1) 90.68(9), O(1s)–Ru(1)–P(1) 167.62(8), N(1)–Ru(1)–P(2) 100.07(9), N(2)–Ru(1)–P(2) 175.09(9), O(1s)–Ru(1)–P(2) 93.91(8), P(1)–Ru(1)–P(2) 94.23(4), N(1)–Ru(1)–Cl(1) 163.42(9), N(2)–Ru(1)–Cl(1) 91.95(9), O(1s)–Ru(1)–Cl(1) 83.24(8), P(1)–Ru(1)–Cl(1) 87.64(4), P(2)–Ru(1)–Cl(1) 88.61(4).
- Marvin was used for drawing and displaying chemical structures and reactions, chemAxon, Marvin 15.2.9.0, 2015, http://www.chemaxon.com Search PubMed.
- M. C. R. Monteiro, F. B. Nascimento, E. M. A. Valle, J. Ellena, E. E. Castellano, A. A. Batista and S. P. Machado, J. Braz. Chem. Soc., 2010, 21(10), 1992 CrossRef CAS.
- H. Wei, J. Yin and E. Wang, Anal. Chem., 2008, 80, 5635 CrossRef CAS PubMed.
- A. N. Shipway, M. Lahav, R. Gabai and I. Willner, Langmuir, 2000, 16, 8789 CrossRef CAS.
- M. Lahav, A. N. Shipway and I. Willner, J. Chem. Soc., Perkin Trans. 1, 1999, 2, 1925 RSC.
- Y. Yang, S. Matsubara, M. Nagomi and J. Shi, Mater. Sci. Eng., B, 2007, 140, 172 CrossRef CAS.
- H. E. Toma, V. M. Zamarion, S. H. Toma and K. Araki, J. Braz. Chem. Soc., 2010, 21, 1158 CrossRef CAS.
- C. S. Weisbecker, M. V. Merritt and G. M. Whitesides, Langmuir, 1996, 12, 3763 CrossRef CAS.
- M. G. Bellino, E. J. Calvo and G. Gordillo, Phys. Chem. Chem. Phys., 2006, 6, 424 RSC.
- G. Von Poelhsitz, A. L. Bogado, L. M. Lião, A. G. Ferreira, E. E. Castellano, J. Ellena and A. A. Bastista, Polyhedron, 2010, 29, 280 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- Enraf-Nonius, Collect, Nonius BV, Delft, The Netherlands, 1997-2000 Search PubMed.
- Z. Otwinowski and W. Minor, HKL Denzo and Scalepack, in Methods in Enzymology, ed. C. W. Carter Jr and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- P. Coppens, L. Leiserowitz and D. Rabinovich, Acta Crystallogr., 1965, 1035 CrossRef CAS.
- G. Frens, Nature (London), Phys. Sci., 1973, 241, 20 CrossRef CAS.
- M. B. Egorova, A. V. Dobrachenkc and A. M. Popov, Koord. Khim., 1987, 13, 541 CAS.
Footnotes |
| † In memoriam of Prof. Peter Hofmann (Organisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg). |
| ‡ Electronic supplementary information (ESI) available. CCDC 1457016, 1457017 and 1456936. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra05616d |
| § Present address: Universidade Federal de Ouro Preto, Campos Morro do Cruzeiro, CEP 35.400-000 Ourto Preto – MG – Brasil. |
| This journal is © The Royal Society of Chemistry 2016 |