
Efficient photocatalytic hydrogen evolution system by assembling earth abundant NixOy nanoclusters in cubic MCM-48 mesoporous materials†
Abstract
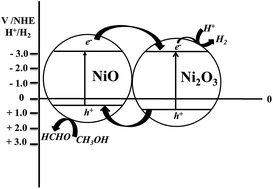
A cubic MCM-48 mesoporous material was employed as a support to encapsulate earth abundant NixOy species (NiO and Ni2O3). The cubic MCM-48 mesoporous support provides an excellent platform to not only effectively disperse NiO and/or Ni2O3 species but also to limit their particle sizes. The presence of Ni2O3 species at an optimal amount seems to enhance the photocatalytic activity of Ni–MCM-48 materials in comparison to a Ni–MCM-48 mesoporous material having only NiO dispersed in it. In addition, the presence of bulk NiO species also seems to be detrimental to the generation of solar hydrogen. The apparent quantum yield (AQY) of the most active material, Ni–MCM-48-2.5% was estimated to be 5.35%. This was over 250 times higher than a bulk, NiO (AQY = 0.02%) under identical experimental conditions. This study indicates that MCM-48 can be used as an effective support to disperse NixOy species.
 Please wait while we load your content...
Please wait while we load your content...

