
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone-catalyzed aerobic oxidation reactions via multistep electron transfers with iron(II) phthalocyanine as an electron-transfer mediator†
Yiming Honga,
Tiantian Fangb,
Meichao Lia,
Zhenlu Shen*a,
Xinquan Hu*a,
Weimin Moa,
Baoxiang Hua,
Nan Suna and
Liqun Jina
aCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: zhenlushen@zjut.edu.cn; xinquan@zjut.edu.cn
bCollege of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, China
First published on 20th May 2016
Abstract
A new biomimetic catalytic oxidation system which employs 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the catalyst, molecular oxygen as the terminal oxidant and iron(II) phthalocyanine (FeIIPc) as the electron-transfer mediator has been developed. This system can be applied for oxidative deprotection of PMB ethers, alcohol oxidation, aromatization and α,β-unsaturated aldehyde formation. After immobilizing FeIIPc on multi-walled carbon nanotubes, it can be reused without loss of activity.
Introduction
Oxidation reactions play an important role in organic synthesis, and numerous oxidants and oxidation methodologies have been developed. Today, there is an increasing demand for the use of molecular oxygen as the terminal oxidant in oxidation reactions due to its remarkable advantages, including great abundance, low cost, and generating water as the only byproduct. This kind of oxidant meets the requirement for sustainable and environmentally benign processes. However, the direct oxidation of common organic compounds by molecular oxygen is difficult because of the high energy barrier in the electron transfer from the organic substrates to molecular oxygen. This is also the reason why the molecular oxygen has a high oxidative potential but shows extraordinary stability. Therefore, oxidation systems which employed substrate-selective catalysts with molecular oxygen as the terminal oxidant have been developed to circumvent the above problem.1 In these systems, the organic substrates can be oxidized by the substrate-selective catalysts, and the reduced catalysts can be reoxidized by molecular oxygen to their active forms.2 However, this approach is not universal, which fails in some cases, due to the electron transfer between catalyst and molecular oxygen is high in energy barrier. Therefore, an alternative method using electron-transfer mediators (ETMs) in the catalysts and molecular oxygen is developed. The ETMs would transfer electrons from catalyst to molecular oxygen along a low-energy pathway (Fig. 1).3 This can be described as a biomimetic approach due to the similarities with biological systems.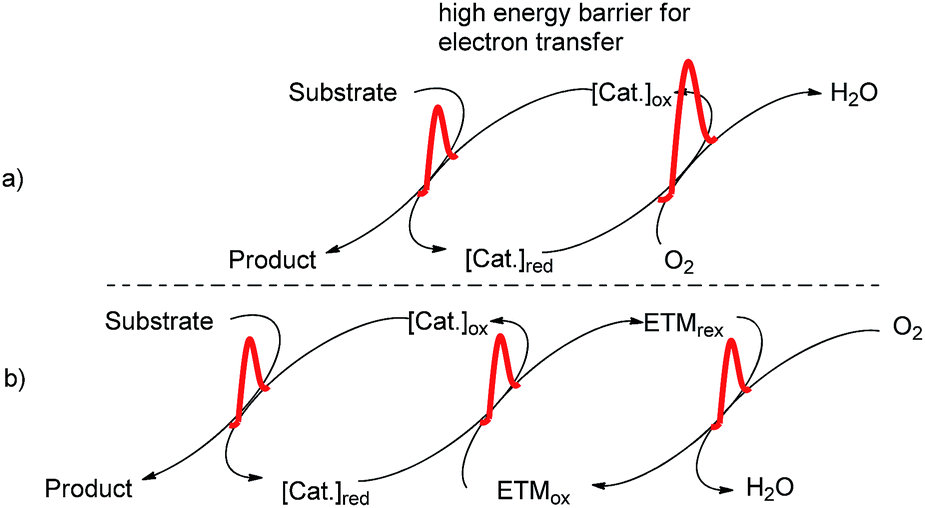 | ||
| Fig. 1 Comparison of (a) direct reoxidation catalysts by O2 and (b) reoxidation catalysts via low-energy electron transfer using ETMs. | ||
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) is a powerful oxidant, and it has been successfully applied in many reactions.4 However, the use of stoichiometric DDQ may result in purification difficulties because of the concomitant by-product 2,3-dichloro-5,6-dicyano-hydroquinone (DDHQ). To avoid this inherent disadvantage, catalytic oxidation systems with DDQ as the catalyst and Mn(OAc)3 (ref. 5) or MnO2 (ref. 6) as the terminal oxidants were developed, while it should be noted that using of a great amount of these inorganic oxidants brought new environmental effluents. We are interested in using molecular oxygen as the terminal oxidant, but it is impossible to directly regenerate DDHQ to DDQ with molecular oxygen, and therefore an additional ETM is needed. Previously, our group developed a DDQ/tert-butyl nitrite (TBN)/O2 catalytic oxidation system for the aerobic oxidation of alcohols, oxidative deprotection of ethers, and oxidation of diarylmethane sp3 C–H bonds,7 which was also utilized by other groups.8 The key point of this aerobic oxidation is that NO released from TBN can act as the ETM between DDQ and molecular oxygen. In fact, this concept was also applied in our other catalytic oxidation systems, such as TEMPO/Br2/NaNO2/O2,9 TEMPO/HBr/TBN/O2,10 TEMPO/TBN/O2 (ref. 11), TEMPO/KPF6/NaNO2/O2,12 and TEMPO/DDQ/TBN/O2 system.13 In addition, azobisisobutyronitrile (AIBN) also can be used as the ETM in the DDQ-catalyzed aerobic oxidations.14
Iron phthalocyanine (FePc) is a macrocyclic compound, whose central iron ion is bonded so strongly that it only can be removed by breaking the macrocycle.15 Having a similar structure with iron porphyrins,16 the efficiencies of FePc as catalyst in many oxidation reactions, such as catalytic oxidation of arenes,17 alkanes,18 phenols,19 olefins20 and other compounds,21 have been demonstrated. Based on the FePc-catalyzed oxidation reactions of hydroquinones to benzoquinones,18 several aerobic oxidation reactions employing palladium as the catalyst, benzoquinone (BQ) as the co-catalysts and FePc as the ETM have been developed.22
Considering that DDQ is a quinone compound, we reasoned that FePc could act as the ETM between DDQ and molecular oxygen. Thus we wish to establish DDQ/FeIIPc/O2 system for the oxidation reactions as described in Scheme 1. This new process involves two redox systems: DDQ/DDHQ–FePcox/FeIIPc. Herein, we reported our research results of this catalytic oxidation system for oxidative deprotection of p-methoxybenzyl (PMB) ethers, alcohol oxidation, aromatization and α,β-unsaturated aldehyde formation. Although DDQ-catalyzed oxidative deprotection of PMB ethers, alcohol oxidation and aromatization using molecular oxygen as the terminal oxidant were reported recently, to the best of our knowledge, this work reported the first example of DDQ-catalyzed conversion of aldehyde to α,β-unsaturated aldehyde using molecular oxygen as the terminal oxidant.
 | ||
| Scheme 1 Proposed DDQ/FeIIPc/O2 catalytic oxidation system. | ||
Results and discussion
By applying it to the oxidative deprotection of PMB ethers, we study the feasibility of the DDQ/FeIIPc/O2 system. In the initial screening experiments, the oxidative deprotection of PMB ether of 2-octanol (1a) was selected as the model reaction (Table 1). Firstly, the oxidative deprotection of 1a was carried out in toluene at 120 °C under 0.5 MPa of oxygen for 24 h. Without DDQ or FeIIPc, the conversion of 1a was poor (entries 1 and 2). In the presence of 20 mol% of FeIIPc and 20 mol% of DDQ, the conversion of 1a was increased to 70%, but the selectivity to 2-octanol (2a) was only 43% (entry 3).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | DDQ (mol%) | FeIIPc (mol%) | Additive (mol%) | T (°C) | t (h) | P (MPa) | Conv.b (%) | Select.b (%) |
| a The reactions were performed by using 1a (1 mmol), and solvent (20 mL).b The conversion and selectivity were determined by GC with area normalization.c GDME: ethylene glycol diethyl ether.d TeCA: 1,1,2,2-tetrachloroethane. | |||||||||
| 1 | Toluene | — | 20 | — | 120 | 24 | 0.5 | 1 | — |
| 2 | Toluene | 20 | — | — | 120 | 24 | 0.5 | 16 | — |
| 3 | Toluene | 20 | 20 | — | 120 | 24 | 0.5 | 70 | 43 |
| 4 | Toluene | 20 | 20 | 2,2′-bpy (20) | 120 | 24 | 0.5 | 70 | 79 |
| 5 | Toluene | 20 | 20 | 4,4′-bpy (20) | 120 | 24 | 0.5 | 100 | 87 |
| 6 | Toluene | 20 | 20 | 4,4′-bpy (20) | 120 | 5 | 0.5 | 100 | 90 |
| 7 | Toluene | 10 | 10 | 4,4′-bpy (10) | 120 | 5 | 0.5 | 100 | 96 |
| 8 | Toluene | 8 | 8 | 4,4′-bpy (8) | 120 | 5 | 0.5 | 86 | 96 |
| 9 | Toluene | 10 | 10 | Pyrazole (10) | 120 | 5 | 0.5 | 52 | 91 |
| 10 | Toluene | 10 | 10 | Imidazole (10) | 120 | 5 | 0.5 | 98 | 92 |
| 11 | Toluene | 10 | 10 | Pyridine (10) | 120 | 5 | 0.5 | 54 | 88 |
| 12 | Toluene | 10 | 10 | 4,4′-bpy (10) | 80 | 5 | 0.5 | 90 | 98 |
| 13 | Toluene | 10 | 10 | 4,4′-bpy (10) | 80 | 5 | 0.4 | 78 | 98 |
| 14 | Toluene | 10 | 10 | 4,4′-bpy (10) | 80 | 6 | 0.4 | 82 | 98 |
| 15 | Toluene | 10 | 10 | 4,4′-bpy (10) | 80 | 8 | 0.4 | 100 | 96 |
| 16 | CH3CN | 10 | 10 | 4,4′-bpy (10) | 80 | 8 | 0.4 | 9 | 98 |
| 17 | DMF | 10 | 10 | 4,4′-bpy (10) | 80 | 8 | 0.4 | 10 | — |
| 18 | GDMEc | 10 | 10 | 4,4′-bpy (10) | 80 | 8 | 0.4 | 25 | — |
| 19 | PhCl | 10 | 10 | 4,4′-bpy (10) | 80 | 8 | 0.4 | 100 | 96 |
| 20 | TeCAd | 10 | 10 | 4,4′-bpy (10) | 80 | 8 | 0.4 | <1 | — |
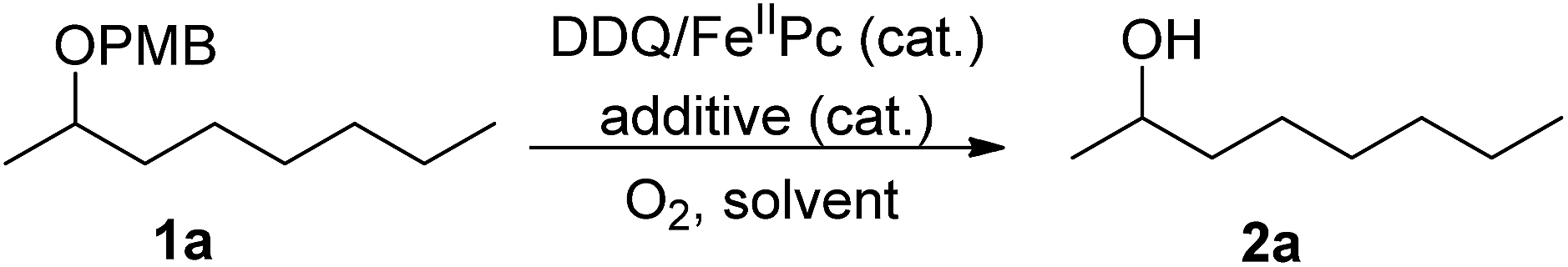
It was well known that nitrogenous ligands could affect the catalytic performance of metalloporphyrin via changing the spin state of the catalyst.23 It also has been reported that nitrogenous compounds could modulate the physicochemical properties of FePc via changing the spin property of the iron atom due to the strong ligand field created by the nitrogenous ligand.15,24 Thus we reasoned nitrogenous compounds could regulate the catalytic performance of FePc, and 2,2′-dipyridyl (2,2′-bpy) was added in our DDQ/FeIIPc/O2 system. To our delight, the selectivity to 2a was dramatically increased to 79%, though the conversion of 1a remained 70% (entry 4). When 2,2′-bpy was replaced by 4,4′-bpy, 100% conversion of 1a with 87% selectivity to 2a was obtained (entry 5). Then the reaction time was shortened, it was found the oxidative deprotection could be completed within 5 h (entry 6). Later on, the loads of DDQ, FeIIPc and 4,4′-bpy were also attempted to be reduced (entries 7 and 8). The experimental results showed that 1a could be fully converted to 2a in 96% selectivity with 10 mol% of FeIIPc, 10 mol% of DDQ and 10 mol% of 4,4′-bpy (entry 7). Other nitrogenous compounds were also screened, 4,4′-bpy gave the best result (entries 7, 9–11).
Cutting down the reaction temperature and pressure of oxygen, the conversion of 1a would be decreased, but not unacceptable (entries 12 and 13). When the reaction time was prolonged to 8 h, the complete conversion of 1a with 96% selectivity to 2a could be achieved at 80 °C under 0.4 MPa of oxygen (entry 15). Among the screened solvents, toluene and chlorobenzene gave the excellent results (entries 15–20). Toluene was selected as the solvent for the next studies because it was more environmentally benign than chlorobenzene.
With the optimized reaction conditions in hand, the results of aerobic oxidative deprotection of a variety of PMB ethers in presence of DDQ, FeIIPc and 4,4′-bpy are summarized in Table 2. All of the substrates could be smoothly converted to their corresponding alcohols. PMB ethers of 1-octanol and cyclohexanol (1b and 1c) underwent a complete deprotection to produce the corresponding alcohols in excellent selectivities (entries 2 and 3). When PMB ethers with high steric hindrance (1d and 1e) were subjected to deprotection, increased catalyst loads or prolonged reaction time were needed (entry 4 and 5). PMB ethers containing a heterocyclic moiety (1f–1i) could also be fully deprotected (entries 6–9). The successful deprotection of 1g showed that the oxidative deprotection can endure the Boc group (entry 7). When mono- or di-PMB protected 1,6-hexanediol (1k and 1k′) were used as the substrates, hexanediol (2k) could be obtained in excellent yields (entries 11 and 12). The substrates with two different hydroxyl protecting groups (1l–1n) were also submitted to the oxidative deprotection. The results showed that the PMB group could be selectively removed to give the corresponding alcohols without affecting the other protecting groups (entries 13–15). Compared with Bn group, PMB group was easy to deprotect. Entry 16 showed that the PMB group was selectively cleaved without affecting the Bn group.
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | t (h) | Conv.b (%) | Select.b (%) | |
| a Reaction conditions: PMB ether (1 mmol), toluene (20 mL), DDQ (10 mol%), FeIIPc (10 mol%), 4,4′-bpy (10 mol%), O2 (0.4 MPa), 80 °C.b The conversion and selectivity were determined by GC with area normalization.c DDQ (15 mol%), FeIIPc (15 mol%), 4,4′-bpy (15 mol%).d DDQ (20 mol%), FeIIPc (20 mol%), 4,4′-bpy (20 mol%). | |||||
| 1 |  |
1a | 8 | 100 | 96 |
| 2c | CH3(CH2)7OPMB | 1b | 14 | 100 | 96 |
| 3 |  |
1c | 10 | 100 | 97 |
| 4c |  |
1d | 20 | 100 | 98 |
| 5 |  |
1e | 14 | 100 | 98 |
| 6d |  |
1f | 24 | 100 | 93 |
| 7c |  |
1g | 18 | 100 | 96 |
| 8 |  |
1h | 16 | 100 | 98 |
| 9 |  |
1i | 8 | 100 | 98 |
| 10c |  |
1j | 10 | 100 | 95 |
| 11 | HO(CH2)6OPMB | 1k | 8 | 100 | 92 |
| 12 | PMBO(CH2)6OPMB | 1k′ | 8 | 100 | 95 |
| 13 | MeO(CH2)6OPMB | 1l | 8 | 100 | 96 |
| 14 | MOMO(CH2)6OPMB | 1m | 16 | 100 | 95 |
| 15 | AcO(CH2)6OPMB | 1n | 8 | 100 | 95 |
| 16 | BnO(CH2)6OPMB | 1o | 8 | 100 | 96 |
To increase the synthetic utility of the reaction, the recyclability of FePc was considered, and the immobilization of FePc on insoluble supports appears to be a good way to make it practicable. Multi-walled carbon nanotubes (MWCNTs) exhibit extraordinary physical and chemical characteristics, and they are good carriers for catalytic applications because of specific metal support interactions given by their graphitic structure as well as specific surface area.25 FePc and Fe–porphyrin could be easily adsorbed on MWCNTs via non-covalent π–π interactions, and immobilization of FePc and Fe–porphyrin on the MWCNTs also have been reported.26 Therefore, the MWCNT-supported FeIIPc (FeIIPc-MWCNT) was prepared and applied to the oxidative deprotection of PMB ethers. Under the reaction conditions of 0.4 MPa of oxygen and toluene as the solvent, the results of oxidative deprotection of PMB ethers are summarized in Table 3. The data in Table 3 show the yields of alcohols to be good to excellent (entries 1–16). Most of the PMB ethers showed high reactivities affording the desired products. Compared with the oxidative deprotection reactions performed with non-supported FeIIPc, most of the reactions performed with FeIIPc-MWNT needed longer reaction time, higher catalyst loads or reaction temperature.
| Entry | Substrate | T (°C) | t (h) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: PMB ether (1 mmol), toluene (20 mL), DDQ (10 mol%), FeIIPc-MWNT (10 mol%), 4,4′-bpy (10 mol%), O2 (0.4 MPa).b Isolated yields, values in parentheses were determined by GC internal standard method.c DDQ (15 mol%), FeIIPc (15 mol%), 4,4′-bpy (15 mol%).d DDQ (20 mol%), FeIIPc (20 mol%), 4,4′-bpy (20 mol%).e O2 (0.5 MPa). | ||||
| 1 | 1a | 80 | 8 | 93 |
| 2c | 1b | 80 | 14 | 87 |
| 3 | 1c | 80 | 10 | 96 |
| 4c | 1d | 80 | 20 | 98 |
| 5c | 1e | 80 | 14 | 82 |
| 6d,e | 1f | 120 | 7 | 65 |
| 7c | 1g | 80 | 18 | 88 |
| 8 | 1h | 80 | 16 | 91 |
| 9d | 1i | 80 | 16 | 78 |
| 10d | 1j | 100 | 10 | 84 |
| 11d | 1k | 120 | 16 | 79 |
| 12d | 1k′ | 120 | 24 | 78 |
| 13d | 1l | 100 | 16 | 71 |
| 14d | 1m | 100 | 16 | 85 |
| 15d | 1n | 100 | 16 | 84 |
| 16d | 1o | 100 | 8 | 91 |
In order to demonstrate the stability and recyclability of FeIIPc-supported, FeIIPc-MWCNT was examined by the oxidative deprotection of 1a under optimized reaction conditions. After completion of each reaction run, FeIIPc-MWCNT could be recovered by simple filtered and washed with toluene. The recovered catalyst was then successfully used in four runs at the expense of a slight decline in catalytic performance (Fig. 2).
 | ||
| Fig. 2 Stability and recyclability of FeIIPc-MWCNT in the oxidative deprotection of 1a. | ||
Then DDQ/FeIIPc/O2 system was examined in other aerobic oxidation reactions to explore the generality of the process. Alcohol oxidation, aromatization and α,β-unsaturated aldehyde formation were selected as examples (Scheme 2). Oxidation of alcohol is quite effective, with cinnamyl alcohol (3) being converted to cinnamaldehyde (4) in 90% isolated yield. This DDQ/FeIIPc/O2 system could also be applied to the aromatization, as seen in the conversion of indoline (5) to indole (6). Hayashi et al. reported a practical conversion of aldehydes into α,β-unsaturated aldehydes with a catalytic amount of DDQ and stoichiometric MnO2.27 Herein, our DDQ/FeIIPc/O2 system successfully replaced DDQ/MnO2 system, and 3-phenylpropanal (7) was converted to cinnamaldehyde (4) in 70% isolated yield. DDQ/TBN/O2 system was also applied to this α,β-unsaturated aldehyde formation reaction, but no desired product could be obtained.
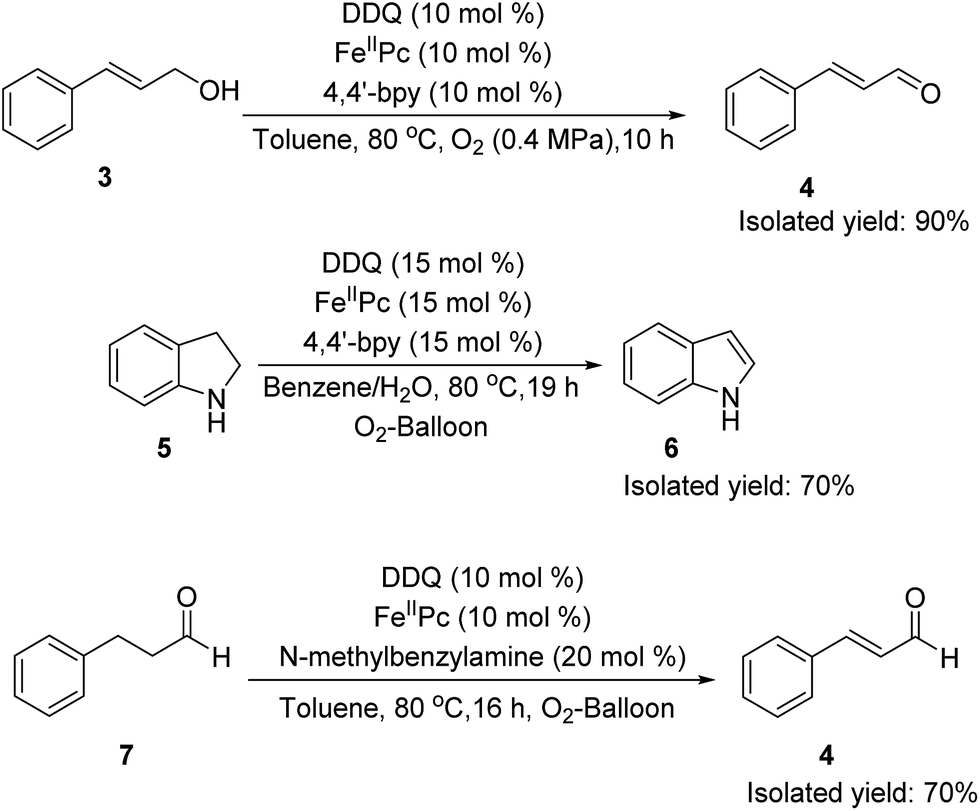 | ||
| Scheme 2 DDQ/FeIIPc-catalyzed aerobic oxidation reactions. | ||
Conclusions
In conclusion, a new catalytic oxidation system has been developed, which employed DDQ as the catalyst, molecular oxygen as the environmentally benign terminal oxidant, FeIIPc as the ETM and 4,4′-bpy as the additive. This system was successfully applied for oxidative deprotection of PMB ether, alcohol oxidation, aromatization and transformation of aldehydes into α,β-unsaturated aldehydes. Under the optimal reaction conditions, a variety of PMB ethers can be deprotected to their corresponding alcohols in excellent conversions and selectivities. For the purpose of reuse of FeIIPc, the MWCNT-supported FeIIPc (FeIIPc-MWCNT) was prepared, which also exhibited good activity in the DDQ-catalytic aerobic oxidative deprotection of PMB ethers. FeIIPc-MWCNT can be easily recovered and reused for at least four times without affecting its catalytic performance.Experimental section
General procedure for oxidative deprotection of PMB ethers with DDQ/FeIIPc/O2 system (Table 2, entry 1)
To a Teflon-lined 316L stainless steel autoclave (300 mL) was added 1 mmol PMB ether of 1a, 10 mol% of DDQ, 10 mol% of FePc, 10 mol% of 4,4′-bpy and 20 mL of toluene. Then closed the autoclave and charged O2 to 0.4 MPa. Put the autoclave into an oil bath, which was preheated to 80 °C. After the reaction was finished, the autoclave was cooled to room temperature and carefully depressurized. Diluted the sample with CH3CN and detected the conversion and selectivity by GC without any purification.General procedure for oxidative deprotection of PMB ethers with DDQ/FeIIPc-MWCNT/O2 system (Table 3, entry 8)
Except that FePc was replaced by FeIIPc-MWCNT, the experimental process was as same as the above description. After the reaction was finished, the reaction mixture was filtered, and FeIIPc-MWCNT was washed with toluene. Then the filtrate was concentrated by rotary evaporator and the residue was purified by column chromatography on silica gel to afford 2h in 91% yield as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.39 (d, J = 8.10 Hz, 1H), 7.15 (d, J = 8.05 Hz, 1H), 4.68–4.70 (t, 1H), 3.61 (s, 1H), 2.83–2.72 (m, 2H), 2.24–2.23 (m, 1H), 2.02–2.00 (m, 1H), 1.85–1.80 (m, 2H).Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21376224, 21206147) and Hangzhou Qianjiang Distinguished Experts Project.Notes and references
- J. Piera and J.-E. Backvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- (a) L. L. Joyce and R. A. Batey, Org. Lett., 2009, 11, 2792–2795 CrossRef CAS PubMed; (b) A. Acharya, S. V. Kumar and H. Ila, Chem.–Eur. J., 2015, 21, 17116–17125 CrossRef CAS PubMed; (c) S. H. Zhang, P. C. Qian, M. L. Zhang, M. L. Hu and J. Cheng, J. Org. Chem., 2010, 75, 6732–6735 CrossRef CAS PubMed.
- (a) B. W. Purse, L.-H. Tran, J. Piera, B. Akermark and J.-E. Backvall, Chem.–Eur. J., 2008, 14, 7500–7503 CrossRef CAS PubMed; (b) S. Luo, F. X. Luo, X. S. Zhang and Z. J. Shi, Angew. Chem., Int. Ed., 2013, 52, 10598–10601 CrossRef CAS PubMed; (c) J.-E. Backvall, R. B. Hopkins, H. Grennberg, M. M. Mader and A. K. Awasth, J. Am. Chem. Soc., 1990, 112, 5160–5166 CrossRef; (d) Z. K. Wickens, K. Skakuj, B. Morandi and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 890–893 CrossRef CAS PubMed; (e) A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638–14658 CrossRef CAS PubMed.
- (a) B. A. McKittrick and B. Ganem, J. Org. Chem., 1985, 50, 5897–5898 CrossRef CAS; (b) B. V. Rokade, S. K. Malekar and K. R. Prabhu, Chem. Commun., 2012, 48, 5506–5508 RSC; (c) Y. H. Jang and S. W. Youn, Org. Lett., 2014, 16, 3720–3723 CrossRef CAS PubMed; (d) Z. M. Wang, H. J. Mo, D. P. Cheng and W. L. Bao, Org. Biomol. Chem., 2012, 10, 4249–4255 RSC.
- (a) G. V. M. Sharma, B. Lavanya, A. K. Mahalingam and P. R. Krishna, Tetrahedron Lett., 2000, 41, 10323–10326 CrossRef CAS; (b) C. C. Cosner, P. J. Cabrera, K. M. Byrd, A. M. Thomas and P. Helquist, Org. Lett., 2011, 13, 2071–2073 CrossRef CAS PubMed.
- (a) H. Yi, Q. Liu, J. Liu, Z. Q. Zeng, Y. H. Yang and A. W. Lei, ChemSusChem, 2012, 5, 2143–2146 CrossRef CAS PubMed; (b) L. Liu and P. E. Floreancig, Org. Lett., 2010, 12, 4686–4689 CrossRef CAS PubMed.
- (a) Z. L. Shen, J. L. Dai, J. Xiong, X. J. He, W. M. Mo, B. X. Hu, N. Sun and X. Q. Hu, Adv. Synth. Catal., 2011, 353, 3031–3038 CrossRef CAS; (b) Z. L. Shen, L. L. Sheng, X. C. Zhang, W. M. Mo, B. X. Hu, N. Sun and X. Q. Hu, Tetrahedron Lett., 2013, 54, 1579–1583 CrossRef CAS; (c) J. Q. Ma, Z. M. Hu, M. C. Li, W. J. Zhao, X. Q. Hu, W. M. Mo, B. X. Hu, N. Sun and Z. L. Shen, Tetrahedron, 2015, 71, 6733–6739 CrossRef CAS.
- (a) K. Walsh, H. F. Sneddon and C. J. Moody, Org. Lett., 2014, 16, 5224–5227 CrossRef CAS PubMed; (b) C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed; (c) K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS PubMed.
- R. H. Liu, X. M. Liang, C. Y. Dong and X. Q. Hu, J. Am. Chem. Soc., 2004, 126, 4112–4113 CrossRef CAS PubMed.
- Y. Xie, W. M. Mo, D. Xu, Z. L. Shen, N. Sun, B. X. Hu and X. Q. Hu, J. Org. Chem., 2007, 72, 4288–4291 CrossRef CAS PubMed.
- X. J. He, Z. L. Shen, W. M. Mo, N. Sun, B. X. Hu and X. Q. Hu, Adv. Synth. Catal., 2009, 351, 89–92 CrossRef CAS.
- C. J. Fang, M. C. Li, X. Q. Hu, W. M. Mo, B. X. Hu, N. Sun, L. Q. Jin and Z. L. Shen, Adv. Synth. Catal., 2016, 358, 1157–1163 CrossRef CAS.
- (a) C. B. Qiu, L. Q. Jin, Z. L. Huang, Z. L. Tang, A. W. Lei, Z. L. Shen, N. Sun, W. M. Mo, B. X. Hu and X. Q. Hu, ChemCatChem, 2012, 4, 76–80 CrossRef CAS; (b) Z. L. Shen, M. Chen, T. T. Fang, M. C. Li, W. M. Mo, B. X. Hu, N. Sun and X. Q. Hu, Tetrahedron Lett., 2015, 56, 2768–2772 CrossRef CAS.
- (a) K. Alagiri, P. Devadig and K. R. Prabhu, Chem.–Eur. J., 2012, 18, 5160–5164 CrossRef CAS PubMed; (b) D. P. Cheng, K. Yuan, X. Y. Zhou and J. Z. Yan, J. Chem. Res., 2012, 38, 751–753 CrossRef.
- C. Isvoranu, B. Wang, E. Ataman, K. Schulte, J. Knudsen, J. N. Andersen, M.-L. Bocquet and J. Schnad, J. Phys. Chem. C, 2011, 115, 20201–20208 CAS.
- (a) S. Rismayanil, M. Fukushima, A. Sawada, H. Ichikawa and K. Tatsumi, J. Mol. Catal. A: Chem., 2004, 217, 13–19 CrossRef; (b) J. W. Huang, W. Z. Huang, W. J. Mei, J. Liu, S. G. Hu and L. N. Ji, J. Mol. Catal. A: Chem., 2000, 156, 275–278 CrossRef CAS.
- (a) H. M. Neu, M. S. Yusubov, V. V. Zhdankin and V. N. Nemykin, Adv. Synth. Catal., 2009, 351, 3168–3174 CrossRef CAS; (b) O. V. Zalomaeva, A. B. Sorokin and O. A. Kholdeeva, Green Chem., 2010, 12, 1076–1082 RSC; (c) E. V. Kudrik and A. B. Sorokin, Chem.–Eur. J., 2008, 14, 7123–7126 CAS.
- (a) A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC; (b) N. Grootboom and T. Nyokong, J. Mol. Catal. A: Chem., 2002, 179, 113–123 CrossRef CAS.
- (a) A. Sorokin and B. Meunier, Chem.–Eur. J., 1996, 2, 1308–1317 CrossRef CAS; (b) T. Schmidt, W. Hartung and F. Wasges, Inorg. Chim. Acta, 1998, 274, 126–129 CrossRef CAS; (c) E. T. Saka and Z. Biyiklioglu, Appl. Organomet. Chem., 2015, 29, 392–399 CrossRef CAS.
- (a) A. B. Sorokin and E. V. Kudrik, Catal. Today, 2011, 159, 37–46 CrossRef CAS; (b) E. K. Leggans, T. J. Barker, K. K. Duncan and D. L. Boger, Org. Lett., 2012, 14, 1428–1431 CrossRef CAS PubMed.
- (a) F. Wang, G. J. Wang, W. T. Sun, T. B. Wang and X. T. Chen, Microporous Mesoporous Mater., 2015, 217, 203–209 CrossRef CAS; (b) M. Bala, P. Verma, U. Sharma, N. Kumar and B. Singh, Green Chem., 2013, 15, 1687–1693 RSC.
- (a) J. Piera, K. Narhi and J.-E. Backvall, Angew. Chem., Int. Ed., 2006, 45, 6914–6917 CrossRef CAS PubMed; (b) N. Gigant and J.-E. Backvall, Chem.–Eur. J., 2013, 19, 10799–10803 CrossRef CAS PubMed; (c) B. P. Babu, X. Meng and J.-E. Backvall, Chem.–Eur. J., 2013, 19, 4140–4145 CrossRef CAS PubMed.
- L. Qiang and G. C. Cheng, Sci. China: Chem., 2012, 55, 2036–2053 CrossRef.
- (a) J. Janczak and R. Kubiak, CrystEngComm, 2010, 12, 3599–3606 RSC; (b) J. Pohmer, M. Hanack and J. O. Barcina, J. Mater. Chem., 1996, 6, 957–962 RSC; (c) M. Sumimoto, Y. Kawashima, K. Hori and H. Fujimoto, Dalton Trans., 2009, 5737–5746 RSC.
- (a) Z. W. Yang, X. Q. Xu, T. J. Li, N. N. Zhang, X. Zhao, W. L. Chen, X. X. Liang, X. L. He and H. C. Ma, Catal. Lett., 2015, 145, 1955–1960 CrossRef CAS; (b) L. Mohammadi-Behzad, M. B. Gholivand, M. Shamsipur, K. Gholivand, A. Barati and A. Gholami, Mater. Sci. Eng., C, 2016, 60, 67–77 CrossRef CAS PubMed; (c) M. Amoli-Diva, K. Pourghazi and S. Hajjaran, Mater. Sci. Eng., C, 2016, 60, 30–36 CrossRef CAS PubMed; (d) C. H. Liu, J. Liu, Y. Y. Zhou, X. L. Cai, Y. Lu, X. Gao and S. D. Wang, Carbon, 2015, 94, 295–300 CrossRef CAS.
- (a) A. Morozan, S. Campidelli, A. Filoramo, B. Jousselme and S. Palacin, Carbon, 2011, 49, 4839–4847 CrossRef CAS; (b) A. Rezaeifard and M. Jafarpour, Catal. Sci. Technol., 2014, 7, 1960–1969 RSC; (c) S. A. Mamuru and K. Ozoemena, Electroanalysis, 2010, 22, 985–994 CrossRef CAS.
- Y. Hayashi, T. Itoh and H. Ishikawa, Adv. Synth. Catal., 2013, 355, 3661–3669 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data and copies of 1H NMR spectra. See DOI: 10.1039/c6ra08921f |
| This journal is © The Royal Society of Chemistry 2016 |