DOI:
10.1039/C6RA08904F
(Paper)
RSC Adv., 2016,
6, 69966-69972
Bio-inspired natural polyphenol cross-linking poly(vinyl alcohol) films with strong integrated strength and toughness†
Received
7th April 2016
, Accepted 29th June 2016
First published on 4th July 2016
Abstract
Polymer composites exhibit very high strength but this is usually found to be at the expense of toughness, which greatly limits their application in many fields. Inspired by the hierarchical architectures in spider silk, we attempted to combine simultaneous high strength and toughness via a hydrogen bonding network. PVA composites with tannic acid (TA) molecules were fabricated by a green and easy-to-scale-up water casting method. The results showed that the hydrogen bonding crosslinking between PVA and TA has strong effect on the Tg, crystallinity, thermal stability and mechanical properties of PVA. In addition, the PVA composite films with 3 wt% TA exhibited, simultaneously, the best tensile strength and toughness, improving by 46% and 27% compared to the pure PVA, respectively.
1. Introduction
Recently, much work on polymeric materials has been focused on designing novel lightweight materials with ultra-high mechanical properties for industrial and commercial applications.1,2 Poly(vinyl alcohol) (PVA), a water-soluble polymer, exhibits excellent biocompatibility, chemical stability and film forming properties, which makes it an ideal polymer matrix material.3 Many studies have indicated that PVA can be reinforced by various reinforcing nanoparticles, such as TiO2,4 graphene,5 CNTs6 and nanodiamond.7 However, it is frequently observed that the strength and stiffness of polymer materials are significantly enhanced by nanofillers, but this is nearly always accompanying a reduced ductility or elongation at break.5,7–9 When it comes to material choice, in particular for the service life and safety of materials, the ductility is a major consideration.10 Therefore, it is still a great challenge to combine good toughness with admirable strength and stiffness.
Nature has found a way to solve the conflict between the strength, stiffness, and toughness and obtain integrated materials by natural materials having complex multiscale hierarchical architectures.2,11–13 In particular, spider silk with hierarchical structures is a representative natural material. Many researchers have shown that highly organized antiparallel β-sheet nanocrystals in spider silk, based on an extensive network of hydrogen bonding interactions, plays a crucial role in storing and dissipating mechanical energy, which makes it one of the strongest, most extensible and toughest materials known.14–17 The interactions of hydrogen bonding are relatively weak compared to covalent bonding, but their dense clustering may extensively be formed via an assembly, resulting an exponential increasing of interfacial interactions, accompanying the sharp improvement of mechanical properties.18–20 Actually, Peng et al. have achieved molecular composites of PVA and poly(p-sulfophenylene terephthalamide) (sPPTA), in which the hydrogen-bonding assembly between PVA and sPPTA leads to the improvement of strength and toughness.21 Although the toughness of PVA/sPPTA composite films is much higher than those reported in the other rigid–rod polymer composites, this value is far less than that of pure PVA films, and therefore can’t meet the requirements of high toughness materials. Therefore, further effort is necessary for balancing both the strength and toughness of materials via a hydrogen bonding assembly.
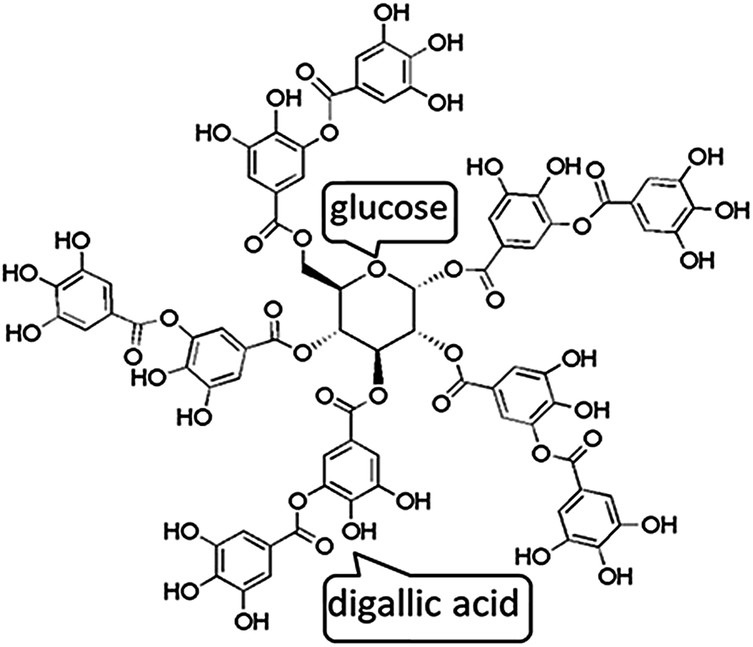
Tannic acid (TA), a high-molecular-weight natural polyphenol that widely exists in the bark and fruits of many plants, is composed of five digallic acid units attached to a central glucose core (Fig. 1). Moreover, TA possesses high biological activity including antioxidant, antimicrobial, antibacterial, antimutagenic, and anticarcinogenic properties, making it widely used for diverse applications.22,23 Recently, TA has drawn much attention for forming intermolecular networks cross-linked by hydrogen bonding with a variety of polymers, such as PEG,24 poly(N-vinylpyrrolidone) (PVP),25 poly(N-isopropylacrylamide) (PNIPAM),26 poly(N-vinylpyrrolidone) (PVPON)27 etc. Thus, TA can develop a stable assembly structure via hydrogen bonding interactions, which has been considered as an effective strategy to prepare the advanced composites.
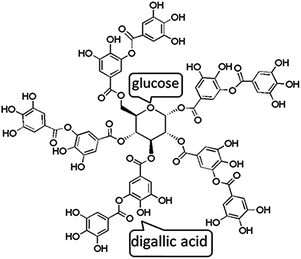 |
| | Fig. 1 Chemical structure of tannic acid. | |
Motivated by the above considerations, we proposed a feasible method for the reinforcement of PVA by TA, which has not been reported so far. We prepared PVA films containing TA molecules by a simple and green water solution blending method. Furthermore, the great number of hydroxy groups on the surface of TA as well as in the PVA molecules would promote the formation of a hydrogen bonding network between the molecule interfaces. In this system, we attempted to comprehensively investigate the influence of hydrogen bonding interactions between TA and PVA on the crystallization behavior, thermal stability, and mechanical properties of the PVA/TA films by varying the ratio of PVA to TA. The hydrogen bonding interactions was commonly characterized by Fourier Transform Infrared (FTIR) spectra. The results indicated that the crosslinking density of hydrogen bonding had a tremendous effect on the crystallinity, thermal stability and mechanical properties.
2. Experimental section
2.1 Materials
PVA was 99% hydrolyzed with an average polymerization degree of 1700 (SINOPEC Sichuan vinylon works) and used as received. TA was purchased from Aladdin Chemical Co., Ltd. The materials were directly used without further purification. All the water was ultrapure water.
2.2 Fabrication of composite films
PVA/TA composite films were prepared by a solvent casting method (Fig. 3A). First, a certain amount of TA was dissolved in ultrapure water by sonication. Meanwhile, 4 g of PVA was dissolved in 34 g of ultrapure water by heating to 90 °C for 2 h to prepare a 10.5 wt% of PVA solution. Then, the TA aqueous solution was gradually added into the PVA solution and continuously stirred for 0.5 h (Fig. 3B). After, the blend was cast onto a glass plate followed by slowly air drying at ambient temperature for 12 h. Finally, polymer films were peeled off and dried under vacuum to a constant weight at 40 °C for 12 h. It should be pointed out that the temperature was maintained at 40 °C instead of higher temperature to avoid any undesired changes of microstructure and hydrogen bonding interactions during the drying process. The as-prepared films were slightly brown and transparent and gradually became dark in color with increased TA content, as shown in Fig. 2. The thickness of the films was about 50–60 μm. The PVA/TA composites obtained with addition content of 1, 3, 5, 7 and 10 wt% of TA are denoted as PVA-1% TA, PVA-3% TA, PVA-5% TA, PVA-7% TA, and PVA-10% TA, respectively.
 |
| | Fig. 2 The digital pictures of the films with various TA contents. | |
 |
| | Fig. 3 (A) Schematic illustration of the interactions for PVA and TA via a simple solution mixing method. (B) Digital images of PVA and the TA blend. (C) Possible hydrogen bonding interactions among the PVA chains and TA molecule. | |
2.3 Characterization and instruments
FTIR spectra of TA and all the composites films were recorded on a Nicolet (Madison, WI, USA) 560 FT-IR spectrophotometer using an attenuated total reflectance (ATR) mode to obtain spectra of high resolution in the range of 500–4000 cm−1. Wide-angle X-ray diffraction (XRD) patterns were measured by RIGAKU SMARTLAB3. Monochromatic CuKa radiation from a rotating anode X-ray generator operating at 40 kV and 30 mA was used. Samples were scanned over the range of diffraction angles 2θ = 3–45°, with a scan speed of 0.5° min−1 at room temperature. Differential scanning calorimetry (DSC) measurements were performed under dry nitrogen using a TA Q200 instrument (TA Instruments) in the temperature range from 30 to 245 °C at 10 °C min−1. Thermogravimetric analysis (TGA) was conducted on a Netzsch TG 209 F1 thermal analyzer at a heating rate of 10 °C min−1 in nitrogen with a temperature range of 30–800 °C. Tensile tests of the films were measured using an Instron 5567 universal testing machine equipped with a 1 kN load cell and each sample was tested at a loading rate of 5 mm min−1 with a gauge length of 30 mm. Because the mechanical properties of PVA and its composites strongly depend on the content of residual water as well as the temperature and humidity at which the tensile strength measurement was conducted, all of the specimens were dried under vacuum to a constant weight at 40 °C for 6 h and conditioned in air at 23 ± 2 °C under a 45% relative humidity (RH) until the moisture equilibrium was reached. All of the samples were cut into strips with a length of 60 mm and a width of 10 mm. The reported values were calculated as averages of over five specimens for each group of specimens. The toughness values of the composites were determined as the area surrounded by the stress–strain curves according to the method reported in the literature.7 Dynamic mechanical analysis (DMA) tests were carried out on a TAQ800 dynamic mechanical analyzer (DMA, USA). The test temperature range was from 20 to 120 °C with a heating rate of 3 °C min−1 and a test frequency of 1 Hz. The water resistance properties of the PVA nanocomposites were evaluated by measuring the water contact angles, the weight swelling ratio and the weight loss from a water-dipping test. The details are shown in the ESI.†
3. Results and discussion
3.1 Fourier transform infrared spectra
In order to investigate whether there were hydrogen bonding interactions between the PVA chains and TA molecules, FTIR was carried out to gain insight into the vibrations of molecules. It is well known that the sharp absorption peak of hydroxyl groups located at 3500–3700 cm−1 corresponds to free hydroxyl groups; that is to say, stretching vibrations of hydroxyl groups appearing at lower wavenumber and showing a wide and a dull peak at 3100–3500 cm−1, reflect the existence of hydrogen bonding interactions.28,29 TA had strong absorption peaks at approximately 3418 cm−1 (Fig. 4) and 1712 cm−1, which were ascribed to the stretching vibrations of phenolic groups (–OH) and carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), respectively. For pure PVA, a broad absorption peak at 3296 cm−1 arose from hydroxyl group (–OH) stretching vibrations owing to the intermolecular and intramolecular hydrogen bonding interactions. The absorption bands at 2941 and 2911 cm−1 were assigned to the methylene group (–CH2–) stretching vibrations of PVA.28 With regard to the PVA/TA films, the peak position of the stretching vibrations of the –OH groups gradually shifted to higher wavenumbers or frequencies with the increase of TA content. The peak position was significantly increased from 3296 cm−1 for pure PVA films to 3318 cm−1 for the PVA-10% TA composite films and obvious blue-shifts indicated new hydrogen bonding (O–H⋯O) being formed between the PVA and TA.30 A complex hydrogen bonding network formed in the PVA/TA system where the phenolic groups of TA and the hydroxyl groups of PVA alternatively served as proton donors or proton acceptors, as shown in Fig. 3C. In summary, the results confirmed the existence of hydrogen bonding interactions between PVA and TA, which was consistent with previous reports on the formation of hydrogen bonding between PVA and melamine or melamine derivatives.31,32
O), respectively. For pure PVA, a broad absorption peak at 3296 cm−1 arose from hydroxyl group (–OH) stretching vibrations owing to the intermolecular and intramolecular hydrogen bonding interactions. The absorption bands at 2941 and 2911 cm−1 were assigned to the methylene group (–CH2–) stretching vibrations of PVA.28 With regard to the PVA/TA films, the peak position of the stretching vibrations of the –OH groups gradually shifted to higher wavenumbers or frequencies with the increase of TA content. The peak position was significantly increased from 3296 cm−1 for pure PVA films to 3318 cm−1 for the PVA-10% TA composite films and obvious blue-shifts indicated new hydrogen bonding (O–H⋯O) being formed between the PVA and TA.30 A complex hydrogen bonding network formed in the PVA/TA system where the phenolic groups of TA and the hydroxyl groups of PVA alternatively served as proton donors or proton acceptors, as shown in Fig. 3C. In summary, the results confirmed the existence of hydrogen bonding interactions between PVA and TA, which was consistent with previous reports on the formation of hydrogen bonding between PVA and melamine or melamine derivatives.31,32
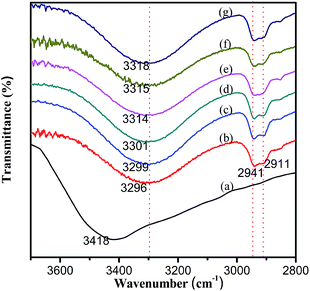 |
| | Fig. 4 FT-IR spectra of the hydroxyl stretching region (a) TA, (b) PVA and PVA/TA composites with different TA content: (c) PVA-1% TA, (d) PVA-3% TA, (e) PVA-5% TA, (f) PVA-7% TA, (g) PVA-10% TA in the range of 3700–2800 cm−1. | |
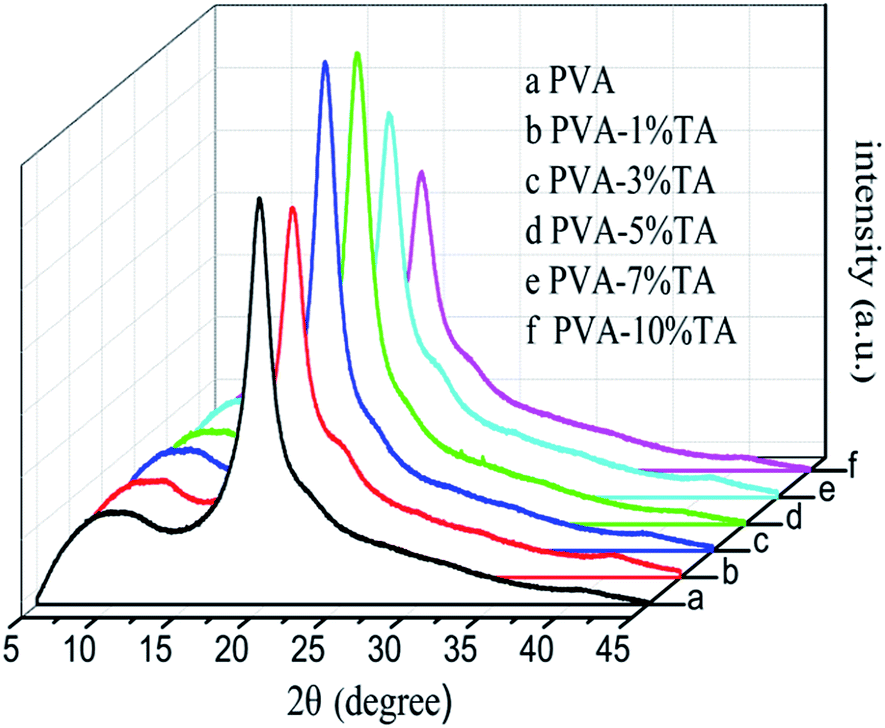
3.2 X-ray diffraction
PVA is a semicrystalline polymer and the degree of crystallinity of the PVA is a crucial factor for its mechanical properties. XRD patterns were used to characterize the effect of the addition of TA content on the crystallinity of PVA (Fig. 5). The XRD pattern of the pure PVA presented a sharp peak at 2θ = 19.6° and a weak shoulder peak at 2θ = 22.5°, corresponding to the (101) and (200) planes of PVA crystals, respectively.33,34 It should be pointed out that the 101 diffraction of PVA crystal is due to the hydrogen bonding within or between the PVA molecular chains. For PVA/TA composites, the TA content did not significantly affect the diffraction peak position, and no new diffraction peaks were discovered. This indicated that the introduction of TA molecules into PVA films didn’t change the crystal structure of PVA. Moreover, the intensity of the diffraction peak at 19.6° of PVA composites increased initially and then decreased with the increase of TA content, demonstrating that the crystallinity of PVA composites had an analogous trend. The results showed that the hydrogen bonding interactions between TA and PVA gradually became stronger with the increase of TA content, which was advantageous to improving the ordering arrangement of PVA molecules. With further additions of TA into PVA, TA can weaken the self-hydrogen bonding of PVA to considerable degree by forming new hydrogen bonding with PVA, and thus restrains the crystallization of PVA and decreases its crystallinity.35 However, PVA-1% TA gave a very low crystallinity, even lower than pure PVA, which possibly indicates that the hydrogen bonding interactions between TA and PVA molecules were too weak, and even can be ignored.
 |
| | Fig. 5 XRD patterns of PVA and PVA/TA composites with varying TA content. | |
3.3 Differential scanning calorimetry
DSC analysis has proved to be a valuable method for researching the effects of hydrogen bonding on the crystallization behavior of PVA/TA composites, as shown in Fig. 6 and in Table 1. The DSC curves of the PVA/TA composites revealed that the thermal behavior of the semi-crystalline polymer had a broad melting peak in the range from 200–240 °C. Apparently, pure PVA exhibited a melting point (Tm) of 233.2 °C and a degree of crystallinity (χc) of 40.5%. Moreover, the melting enthalpy (ΔH0f) of 100% crystalline PVA was regarded empirically as 138.6 J g−1.5,36 When the TA content was increased from 0 to 5%, the melting enthalpy increased, manifesting that the crystallinity also increased. However, PVA-10% TA had a very low χc of 36.2%, even lower than 40.5% for pure PVA. The results also presented an analogous trend with the obtained data by XRD measurements. The results were mainly because at a lower content, TA can induce the crystallization of PVA in view of the appropriate hydrogen bonding crosslinking. But the crystallinity of PVA-1% TA was much higher than that of pure PVA, which was contrary to the data obtained by the DSC tests due to different measurement methods. Apparently, TA, in a higher content, can inhibit the crystallization of PVA due to the formation of a strong physical crosslinking effect. The observed crystallization behaviors are basically consistent with previous literature reports.21 Moreover, the melting point (Tm) and the crystal temperature (Tc) gradually decreased with the addition of TA from 1 to 10%. As for the descent of the melting point, this has a close connection with the crystallinity, but is not entirely determined by the degree of crystallinity.32 Therefore, we can safely draw the conclusion that the hydrogen bonding plays an essential role in the crystallization behavior of the PVA composites.
 |
| | Fig. 6 DSC curves of PVA and PVA/TA composites with varying TA content. | |
Table 1 DSC crystallization and melting enthalpies of PVA and its composites
| Run |
Tm (°C) |
ΔHf (J g−1) |
χc (%) |
Tc (°C) |
| PVA |
233.2 |
56.2 |
40.5 |
197.6 |
| PVA-1% TA |
232.6 |
57.2 |
41.3 |
206.5 |
| PVA-3% TA |
228.6 |
70.3 |
50.7 |
202.0 |
| PVA-5% TA |
227.2 |
75.1 |
52.0 |
201.9 |
| PVA-7% TA |
224.3 |
51.9 |
37.4 |
194.1 |
| PVA-10% TA |
221.1 |
50.2 |
36.2 |
184.5 |
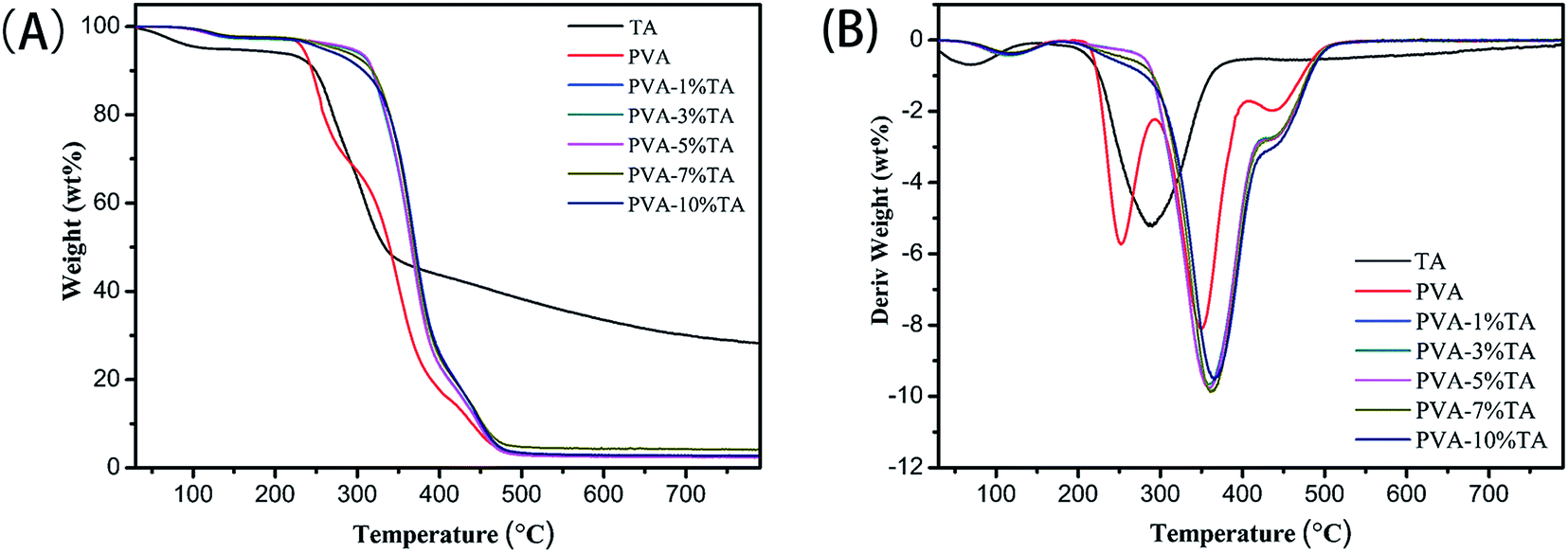
3.4 Thermogravimetric analysis
The TGA and relevant DTG curves presented in Fig. 7 clearly show the thermal stability of PVA and the PVA/TA composites. The initial weight loss from room temperature to 170 °C was attributed to the evaporation of the physically weakly and chemically strongly bound water, and was observed in all samples. After 200 °C, we clearly observed a difference between PVA and the PVA/TA composites. For the pure PVA, the DTG curves showed three distinct peaks corresponding to mass loss associated with three decomposition steps. The first peak (∼252 °C) and the second peak (∼350 °C) can be assigned to the decomposition of the side chains of PVA with the formation of volatile products while the third weak peak (∼435 °C) can be identified as the decomposition of the main chains of PVA.37,38 For the TA, the highest weight loss of about 49% was observed at 170–400 °C.39 As for PVA/TA composites, the initial decomposition temperature increased and the main decomposition peak at about 350 °C shifted to higher temperature with the increasing of TA content. In addition, the corresponding temperature of maximum decomposition rate of PVA-10% TA increased to 366 °C, which was 16 °C beyond the pure PVA. These results can be attributed the formation of hydrogen bonding from hydroxyl groups of PVA and TA, which effectively suppresses the degradation of the side chains of PVA, thus notably improving the thermal stability of the PVA/TA composites.37 Moreover, it was noteworthy that the disappearance of the peak at about 250 °C in the DTG curves meant the inhibition of the dehydration process of –OH functionalities, which also revealed strong physical crosslinking interactions had occurred between PVA and TA. To sum up, the above data intensely indicates that the addition of TA significantly improved the thermal stability of PVA.
 |
| | Fig. 7 (A) TGA and (B) DTG curves of neat PVA and PVA/TA composites with varying TA content. | |
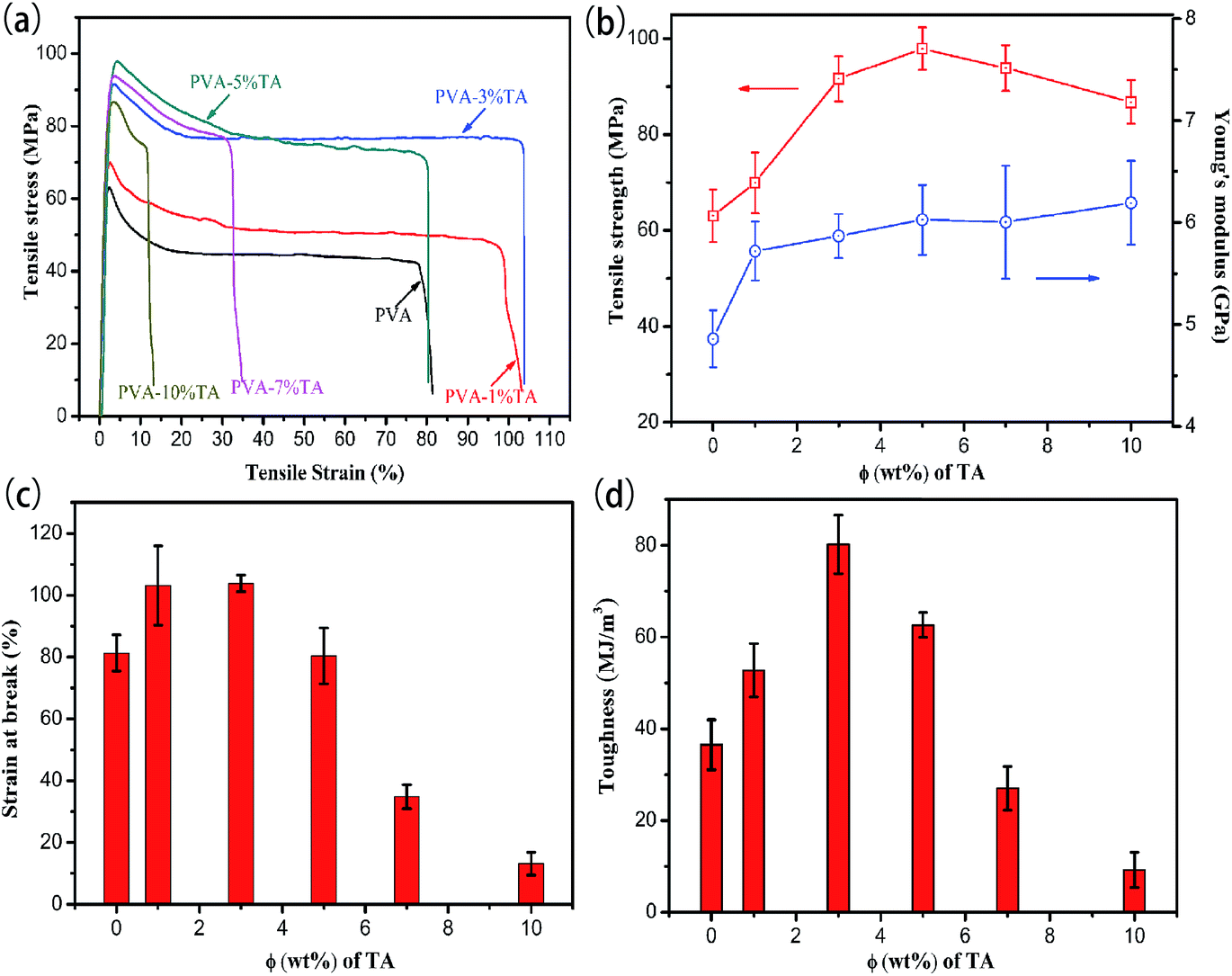
3.5 Mechanical properties
To figure out the reinforcing and toughening effect of TA component on the PVA/TA composite films, tensile mechanical testing was implemented. The typical stress–strain curves, tensile strength, Young’s modules, elongation at break and toughness for PVA and the PVA/TA composite films are presented in Fig. 8. The tensile strength and Young’s modulus of the PVA films increased with the content of TA and reached 86.7 ± 4.5 MPa and 6.2 ± 0.4 GPa, respectively. When the TA content was 5%, the tensile strength reached the maximum value (97.9 ± 4.4) MPa, approximately 55.2% beyond that of pure PVA, suggesting that there were strong intermolecular interactions between PVA and TA. Since TA contains various types of oxygen functional groups, PVA chains can be entangled and/or physically cross-linked with the TA molecule by forming multiple hydrogen bonds, which is desirable for improving the strength.40 However, a further increasing of the TA content brought about a declined tensile strength because TA may play a plasticizing role in the PVA matrix.32 Soares and Soldi proposed that plasticizers reduced the intermolecular interactions and increased the amount of hydrogen bonding.41 Polar groups (–OH) of the plasticizer was believed to form polymer–plasticizer hydrogen bonding, replacing polymer–polymer interactions and hence leading to lower values of tensile strength.
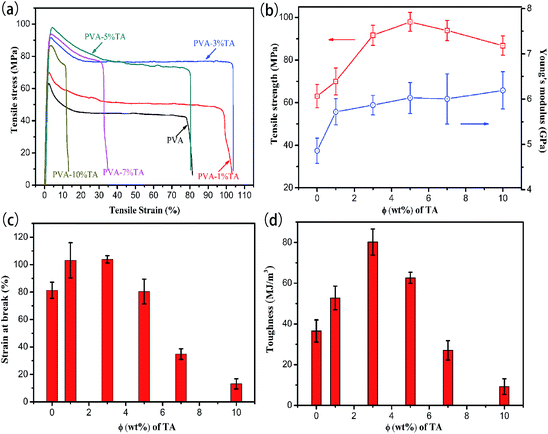 |
| | Fig. 8 Typical stress–strain curves (a), tensile strength values and Young’s modulus (b), and elongation at break (c) and toughness values (d) of PVA/TA films with varying TA content. | |
Interestingly, the PVA composite films also exhibited a large elongation at break (103.9 ± 2.7%) and toughness (80.2 ± 6.3 MJ m−3) with adding 3.0% TA. The improvement of toughness can be explained by the bio-inspired toughening mechanism.42,43 On the one hand, PVA chains can be entangled and/or physically cross-linked on the surface of the TA by forming hydrogen bonding. The entangled and/or physically cross-linked PVA chains can be released, which can dissipate large amounts of energy at the interface between the TA and PVA matrix, leading to the toughening of PVA. On the other hand, the hydrogen bonding network between PVA and TA can be destroyed and rapidly re-built upon the introduction of tension, ensuring that effective load transfer or efficient energy dissipation can exist continuously at the interface between the TA and PVA matrix, which delays the failure of PVA during the tensile process. Furthermore, when the content of TA reached 5% or more, the elongation at break values of PVA/TA decreased significantly, which possibly ascribed to the higher crosslinking density of hydrogen bonding. The composite films became more brittle, indicating that the incorporation of larger amounts of TA increased the degree of entanglement or physical cross-linking of PVA chains, which could restrict the dissociation of interfacial bonding between the TA and PVA, leading to a decrease of the elongation at break and toughness of PVA/TA. In summary, it was obvious that the substantial mechanical improvements are mainly ascribed to the formation of strong multiple hydrogen bonding network.
More importantly, both the ductility and strength of PVA nanocomposites have also aroused researchers’ interest. Generally, strength and toughness are considered to be mutually exclusive.11 For examples, the tensile strength value reported by Seira Morimune et al.7 increased from 95 to 124 MPa (1.3 times) with 5% nanodiamond, while the elongation at break decreased from 72 to 16% (4.5 times), compared with the pure PVA films. Surprisingly, both the tensile strength 91.6 MPa and elongation at break values 103.8% of PVA-3% TA were larger than of pure PVA. Apparently, the introduction of TA molecules into the PVA matrix resulted in distinctly strong and ductile composites, which is valuable for some special applications.
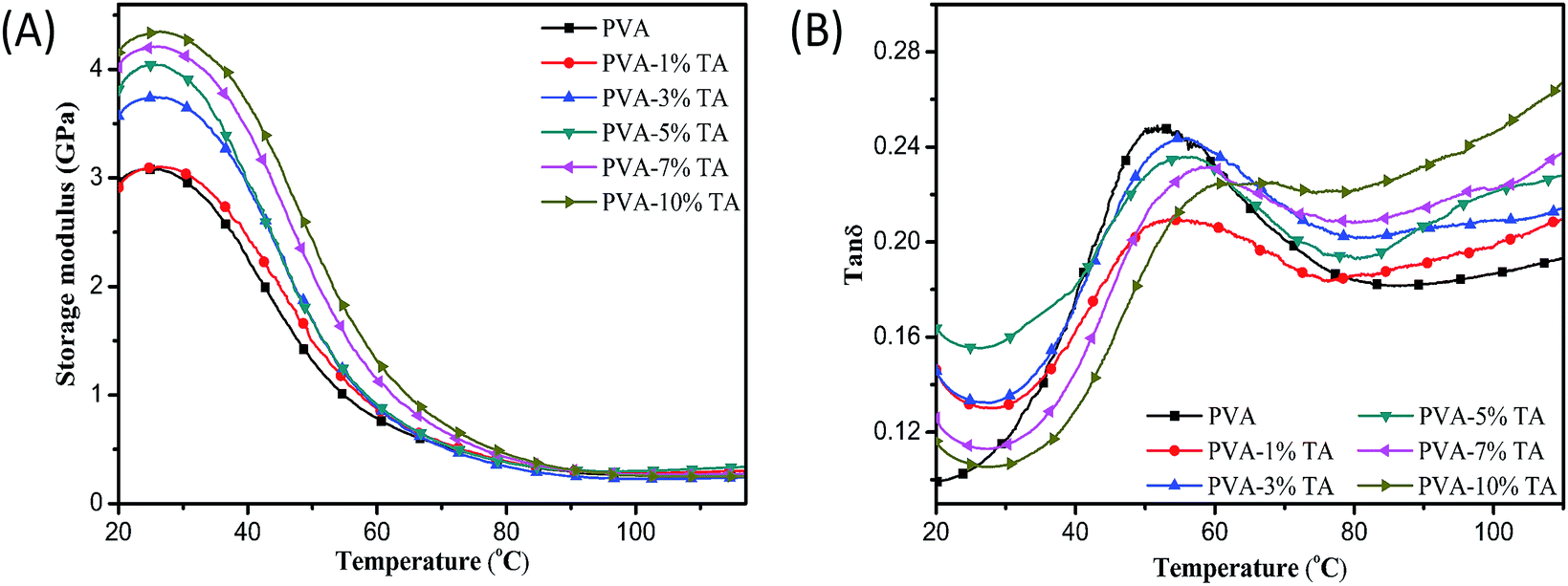
3.6 Dynamic mechanical properties
Dynamic mechanical analysis is one of the most effective and desirable approaches to measure the modulus and loss of material, namely the storage modulus (E′) and loss tangent (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ). Fig. 9 shows plots of the storage modulus and loss tangent versus scan temperature for PVA and PVA/TA composites. The DMA results showed that PVA has a storage modulus of about 3.09 GPa, much lower than that obtained by tensile tests due to different measurement methods. The storage modulus of the PVA/TA films was obviously higher that of pure PVA and increased with the content of TA, which further confirmed that TA was able to enhance the mechanical properties of PVA, because of its strong hydrogen bonding interactions with the PVA matrix. The tanδ peak for pure PVA was observed at approximately 51 °C and represents the Tg of PVA, reflecting the movement state of the microscopic molecular chains. As for PVA/TA films, the tanδ peak obviously shifted to a considerably higher temperature and the peak also became broader. Moreover, the Tg of PVA-10% TA films showed an increase of approximately 12 °C compared with pure PVA. The results demonstrated that the strong interactions of hydrogen bonding restricted the mobility of the PVA chain segments and made the molecular chain tend to be rigid, bringing about the improvement of mechanical properties of the PVA composites.44
δ). Fig. 9 shows plots of the storage modulus and loss tangent versus scan temperature for PVA and PVA/TA composites. The DMA results showed that PVA has a storage modulus of about 3.09 GPa, much lower than that obtained by tensile tests due to different measurement methods. The storage modulus of the PVA/TA films was obviously higher that of pure PVA and increased with the content of TA, which further confirmed that TA was able to enhance the mechanical properties of PVA, because of its strong hydrogen bonding interactions with the PVA matrix. The tanδ peak for pure PVA was observed at approximately 51 °C and represents the Tg of PVA, reflecting the movement state of the microscopic molecular chains. As for PVA/TA films, the tanδ peak obviously shifted to a considerably higher temperature and the peak also became broader. Moreover, the Tg of PVA-10% TA films showed an increase of approximately 12 °C compared with pure PVA. The results demonstrated that the strong interactions of hydrogen bonding restricted the mobility of the PVA chain segments and made the molecular chain tend to be rigid, bringing about the improvement of mechanical properties of the PVA composites.44
 |
| | Fig. 9 (A) Temperature dependence of the storage modulus (E′) and (B) tanδ of pure PVA, PVA/TA composites. | |
4. Conclusions
In summary, natural polyphenol TA reinforced PVA composite films exhibiting excellent tensile strength and fracture toughness were successfully prepared by solution casting and were well characterized. The intermolecular hydrogen bonding interactions in the PVA/TA films were confirmed through the shift of hydroxyl stretching vibration peaks. Importantly, the properties of composites were strongly dependent on the TA content. In addition, the melting point and crystallinity of PVA are obviously increased at a low TA content due to the relatively low density of hydrogen bonding. With the increase of TA content in the films, the strength and ductility of the composites increased up to an optimal value, beyond which mechanical performance decreases in tension. Furthermore, the hydrogen bonding network can also enhance the thermal stability of PVA. We believed that the PVA/TA films with a stable assembly structure via hydrogen bonding interactions have tremendous potential to be used in bio-medicine and biological tissue engineering fields in the future.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 51173115).
Notes and references
- E. Munch, M. E. Launey, D. H. Alsem, E. Saiz, A. P. Tomsia and R. O. Ritchie, Science, 2008, 322, 1516–1520 CrossRef CAS PubMed.
- L. J. Bonderer, A. R. Studart and L. J. Gauckler, Science, 2008, 319, 1069–1073 CrossRef CAS PubMed.
- C. Shao, H. Y. Kim, J. Gong, B. Ding, D. R. Lee and S. J. Park, Mater. Lett., 2003, 57, 1579–1584 CrossRef CAS.
- M. Sairam, M. Patil, R. Veerapur, S. Patil and T. Aminabhavi, J. Membr. Sci., 2006, 281, 95–102 CrossRef CAS.
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS.
- Y. Hou, J. Tang, H. Zhang, C. Qian, Y. Feng and J. Liu, ACS Nano, 2009, 5, 1057–1062 CrossRef PubMed.
- S. Morimune, M. Kotera, T. Nishino, K. Goto and K. Hata, Macromolecules, 2011, 44, 4415–4421 CrossRef CAS.
- P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim, J. D. Xu, H. Nandivada, B. G. Pumplin, J. Lahann and A. Ramamoorthy, Science, 2007, 318, 80–83 CrossRef CAS PubMed.
- X. Zhao, Q. Zhang, D. Chen and P. Lu, Macromolecules, 2010, 43, 2357–2363 CrossRef CAS.
- C. Sanchez, H. Arribart and M. M. G. Guille, Nat. Mater., 2005, 4, 277–288 CrossRef CAS PubMed.
- M. E. Launey and R. O. Ritchie, Adv. Mater., 2009, 11, 2103–2110 CrossRef.
- J. Han, Y. Dou, D. Yan, J. Ma, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2011, 47, 5274–5276 RSC.
- Z. Chen and H. Lu, J. Mater. Chem., 2012, 22, 12479 RSC.
- S. Keten, Z. Xu, B. Ihle and M. J. Buehler, Nat. Mater., 2010, 9, 359–367 CrossRef CAS PubMed.
- A. Nova, S. Keten, N. M. Pugno, A. Redaelli and M. J. Buehler, Nano Lett., 2010, 10, 2626–2634 CrossRef CAS PubMed.
- T. Giesa, M. Arslan, N. M. Pugno and M. J. Buehler, Nano Lett., 2011, 11, 5038–5046 CrossRef CAS PubMed.
- S. Keten and M. J. Buehler, Nano Lett., 2008, 8, 743–748 CrossRef CAS PubMed.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- W. Cui, M. Li, J. Liu, B. Wang, C. Zhang, L. Jiang and Q. Cheng, ACS Nano, 2014, 8, 9511–9517 CrossRef CAS PubMed.
- A. M. Beese, S. Sarkar, A. Nair, M. Naraghi, Z. An, A. Moravsky, R. O. Loutfy, M. J. Buehler, S. T. Nguyen and H. D. Espinosa, ACS Nano, 2013, 7, 3434–3446 CrossRef CAS PubMed.
- M. Peng, G. Xiao, X. Tang and Y. Zhou, Macromolecules, 2014, 47, 8411–8419 CrossRef CAS.
- H. A. Cheng, C. T. Drinnan, N. Pleshko and O. Z. Fisher, Soft Matter, 2015, 11, 7783–7791 RSC.
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Angew. Chem., Int. Ed., 2013, 52, 10766–10770 CrossRef CAS PubMed.
- K. C. Yen and E. M. Woo, Polym. Bull., 2009, 62, 225–235 CrossRef CAS.
- M. Dierendonck, K. Fierens, R. D. Rycke, L. Lybaert, S. Maji, Z. Zhang, Q. Zhang, R. Hoogenboom, B. N. Lambrecht, J. Grooten, J. P. Remon, S. D. Koker and B. G. D. Geest, Adv. Funct. Mater., 2014, 24, 4634–4644 CrossRef CAS.
- E. Costa, M. Coelho, L. M. Ilharco, A. A. Ricardo and P. T. Hammond, Macromolecules, 2011, 44, 612–621 CrossRef CAS.
- V. Kozlovskaya, S. Harbaugh, I. Drachuk, O. Shchepelina, N. K. Loughnane, M. Stone and V. V. Tsukruk, Soft Matter, 2011, 7, 2364–2372 RSC.
- L. Dai and L. Ying, Macromol. Mater. Eng., 2002, 287, 509–514 CrossRef CAS.
- H. S. Mansur, R. L. Oréfice and A. A. P. Mansur, Polymer, 2004, 45, 7193–7202 CrossRef CAS.
- B. Fei, C. Chen, H. Wu, S. Peng, X. Wang and L. Dong, Eur. Polym. J., 2003, 39, 1939–1946 CrossRef CAS.
- P. Song, Z. Xu, Y. Lu and Q. Guo, Macromolecules, 2015, 48, 3957–3964 CrossRef CAS.
- P. Song, Z. Xu and Q. Guo, ACS Macro Lett., 2013, 2, 1100–1104 CrossRef CAS.
- Y. Bin, M. Mine, A. Koganemaru, X. Jiang and M. Matsuo, Polymer, 2006, 47, 1308–1317 CrossRef CAS.
- Z. Zhang, Z. Mo, H. Zhang, X. Wang and X. Zhao, Macromol. Chem. Phys., 2003, 204, 1557–1566 CrossRef CAS.
- J. Li, L. Shao, L. Yuan and Y. Wang, Mater. Des., 2014, 54, 520–525 CrossRef CAS.
- J. Su, Q. Wang, R. Su, K. Wang, Q. Zhang and Q. Fu, J. Appl. Polym. Sci., 2008, 107, 4070–4075 CrossRef CAS.
- N. Wang, L. Zhao, C. Zhang and L. Li, J. Appl. Polym. Sci., 2016, 133, 43246 Search PubMed.
- A. Kaboorani and B. Riedl, Int. J. Adhes. Adhes., 2011, 31, 605–611 CrossRef CAS.
- Z. Xia, A. Singh, W. Kiratitanavit, R. Mosurkal, J. Kumar and R. Nagarajan, Thermochim. Acta, 2015, 605, 77–85 CrossRef CAS.
- M. Y. Lim, H. Shin, D. M. Shin, S. S. Lee and J. C. Lee, Polymer, 2016, 84, 89–98 CrossRef CAS.
- R. M. D. Soares and V. Soldi, Mater. Sci. Eng., C, 2010, 5, 691–698 CrossRef.
- G. E. Fantner, E. Oroudjev, G. Schitter, L. S. Golde, P. Thurner, M. M. Finch, P. Turner, T. Gutsmann, D. E. Morse, H. Hansma and P. K. Hansma, Biophys. J., 2006, 90, 1411–1418 CrossRef CAS PubMed.
- J. Zhang, N. Wang, W. Liu, X. Zhao and W. Lu, Soft Matter, 2013, 9, 6331–6337 RSC.
- E. H. Qua, P. R. Hornsby, H. S. Sharma, G. Lyons and R. D. McCall, J. Appl. Polym. Sci., 2009, 113, 2238–2247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08904f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.