
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Competing and simultaneous click reactions at the interface and in solution†
Doungporn
Yiamsawas
,
Manfred
Wagner
,
Grit
Baier
,
Katharina
Landfester
* and
Frederik R.
Wurm
 *
*
Max-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de; landfester@mpip-mainz.mpg.de
First published on 19th May 2016
Abstract
The kinetics of two simultaneous “click” reactions (thiol–maleimide addition and thiol–disulfide exchange) were investigated by NMR spectroscopy in homogeneous solution and at the oil/water interface of an inverse miniemulsion. For the polyaddition/-condensation of difunctional reagents it was found that the thiol–disulfide exchange is faster than the thiol–maleimide reaction. The addition of a basic catalyst influences the copolymerization behavior of the competitive “click” reactions.
Chemistry for biomedical applications requires modular and specific reactions under mild, aqueous conditions. The thiol–ene reaction is an attractive approach for bio applications, as it can follow “click” characteristics and is inert to most functional groups in biomolecules, but can also selectively be used to address cysteine residues for example.1,2 The thiol–ene reaction has been used for dendrimer synthesis,3 nanoparticle modification,4 or polymer post modification.5 Thiol–ene reactions can proceed via a Michael-type addition (catalyzed by acids, bases,6 or nucleophiles1,7) or by a radical pathway8,9 and they can be conducted in polar solvents such as water, alcohols, or DMF.10 The main highlight of this reaction is a wide range of suitable substrates, including activated and non-activated olefins, as well as multiple-substituted double bonds.2,11 However, all thiol–ene reactions lead to stable thioether linkages. The combination with an another efficient reaction that allows the introduction of cleavable disulfides would be attractive for the design of drug–polymer conjugates or biodegradable nanocarriers. The thiol–disulfide exchange reaction12,13 (due to the reversible cleavage and formation of a new covalent S–S bond) is a powerful tool to be combined with thiol–ene reactions. A recent report from our group demonstrated the successful synthesis of biocompatible DNA-based nanocarriers through the interfacial thiol–disulfide exchange and interfacial thiol–ene reactions in inverse miniemulsion.14 This strategy allowed the combination of an efficient polyaddition and polycondensation with the cleavability of S–S-bonds in biological environment, however, the kinetics of the two concurrent reactions remained unclear.
As both, the thiol–disulfide exchange and the thiol–maleimide “click”, require the same intermediate, i.e. the thiolate anion, their reactions kinetics are essential to understand in competing reactions. The thiol–maleimide reaction requires the initial formation of the thiolate anion.15,16 The mechanism of thiol–disulfide exchange also involves the initial ionization of the thiol to thiolate anion (Scheme 1). To the best of our knowledge, there is no report on the kinetic study of the two competitive “click” reactions: thiol–maleimide addition and thiol–disulfide exchange. Herein, we use difunctional molecules, i.e. a difunctional pyridyldisulfide (1), a dimaleimide (2), and a dithiol (3) to investigate the concurrent polyaddition/-condensation of the three monomers. We study the kinetics of the two reactions both in solution and at the interface of droplets in a water-in-oil miniemulsion. Bucillamine (2) with two thiol groups (pKa 8.39 and 10.22) is selected as a model drug and monomer for the kinetic study. It can both react as a B2-monomer with 1,4-bis-(3-(2-pyridyldithio)propionamido) butane (BPB, 1) or 1,1′-(methylenedi-4,1-phenylene) bismaleimide (2) as the respective A2-monomers (Scheme 2). Since the two thiols of monomer 3 have different reactivities, also the final products include additional structural isomers. First, we investigated the reaction kinetics of both reactions separately in solution. Then, the reactions were investigated in an water-in-oil miniemulsion (i.e. in the presence of a surfactant) with stable aqueous nanodroplets containing 2 and a continuous chloroform phase containing 1 and 3; the reaction takes place at the water–oil interface of the droplets (Scheme 3).
 | ||
| Scheme 1 (a) Thiol–disulfide exchange reaction (with R′′ as a good leaving group, e.g. pyridine-2-thiol); (b) thiol–maleimide reaction (nucleophilic addition). | ||
 | ||
| Scheme 2 Structures of the three difunctional monomers used in this study (A2-monomer: 1,4-bis-(3-(2-pyridyldithio)propionamido) butane (1); A′2-monomer: 1,1′-(methylenedi-4,1-phenylene) bismaleimide (2); B2-monomer: bucillamine (3)) and their reaction pathways. | ||
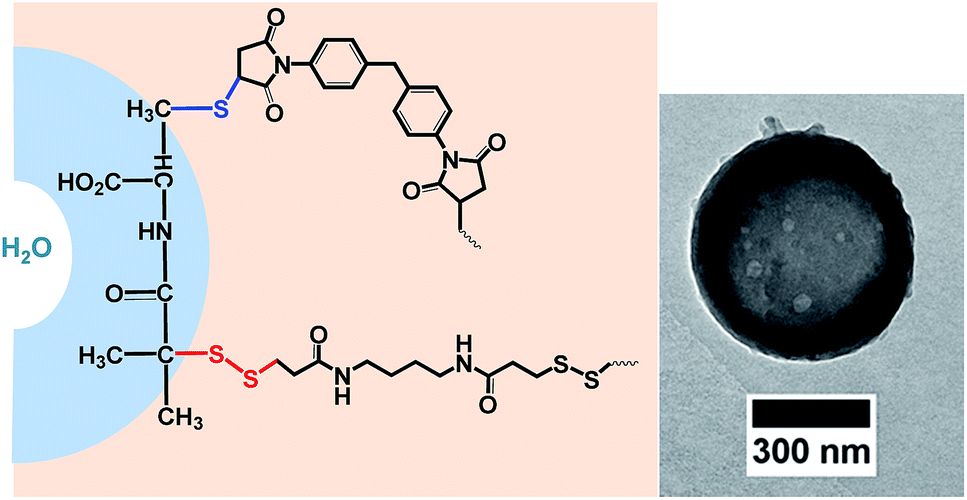 | ||
| Scheme 3 Left: Competitive click reactions between A2 and A′2 with B2 at the interface of a water nanodroplet in chloroform. Right: Representative TEM image of a nanocontainer obtained by interfacial polyaddition/-condensation reaction (after redispersion in water). | ||
In this study, we exploit the advantages of real-time NMR spectroscopy which is well-established method for monitoring reaction kinetics. The use of 1H NMR spectroscopy to study real time polymerization both in solution and inverse miniemulsion polymerization has been previously successful reported from our group for both chain and step growth polymerization.17,18 Herein, the in situ kinetics of both the thiol–disulfide exchange and thiol–maleimide reactions were measured over a period of 600 min. All reactions in solution were carried out in THF-d8 (as a solvent that dissolves all monomers) and in inverse miniemulsion with chloroform-d as the continuous phase and D2O as the dispersed phase. Triethylamine (TEA) was used as a basic catalyst. The reactions were carried out with a delay time of approximately 5 min between each spectral acquisition. In a typical reaction, 2 eq. of bucillamine, 1 eq. maleimide, and 1 eq. disulfide were added into the NMR tube containing 0.75 mL of the deuterated solvent. The evolution of the integrals of specific protons of each reactant was followed over time and compared to an inert internal standard (0.13 ppm from hexamethylcyclotrisiloxane). The maleimide resonance of 2 at 6.9 ppm monitored for the thiol–maleimide and the resonances of the pyridine ring of 1 (at 8.4 ppm) were monitored for the thiol–disulfide interchange, respectively (cf. Fig. S1–S7†).
In thiol–disulfide interchange reactions, a thiol is exchanged with a reactive disulfide, resulting in the formation of a new disulfide by the release of pyridine-2-thiol. This reaction proceeds as a nucleophilic displacement, the thiol nucleophile (thiolate anion) attacking the electrophilic disulfide. The rate of this reaction is dependent on the nucleophilicity of the thiol (RSH – note: in the case of 2 both thiols have a slightly different nucleophilicity which was not considered in all reactions). As a nucleophilic addition, also the thiol–maleimide addition depends on the nucleophilicity of the thiols.
To determine the reaction order of both reactions, the different rate equations for second order reactions can be considered:
 | (1) |
| ln[x]t = −k1t + ln[x]0, n = 1; first-order reaction |

Based on these equations of the rate law, linear plot fitted the best for the first hour to the second-order in the solution system for both the thiol–maleimide reaction and the thiol–disulfide exchange. When the rate constants (k2) of the thiol–maleimide reaction were examined as a function of the catalyst concentration, an increase in the rate was observed as the TEA concentration in the reaction was increased. The reaction between 2 and 3 was accelerated from 0 to 0.0066 min−1 mol−1 L as the amount of TEA was increased from 0 to 0.2 eq. at 298 K. For lower temperatures (283 K) a very similar reaction kinetics were achieved when the amount of TEA was increased to 1 eq. (Fig. 1, top, shows the decrease of 2 measured in solution from 1H NMR kinetics at different temperatures and TEA concentrations). When the same reaction was performed in inverse miniemulsion at 283 K (0.10 mL D2O and 0.65 mL chloroform, c2 = 0.056 mol L−1, c3 = 0.011 mol L−1) the initial reaction speed for different TEA concentrations is increased by a factor of ca. 4 (Fig. 1), however, after a certain period of time the bismaleimide concentration remained constant. This might be due to the formation of an insoluble polymer–shell at the interface of the droplets, i.e. the formation of nanocapsules (cf. Fig. S8†), which terminates the polyaddition.
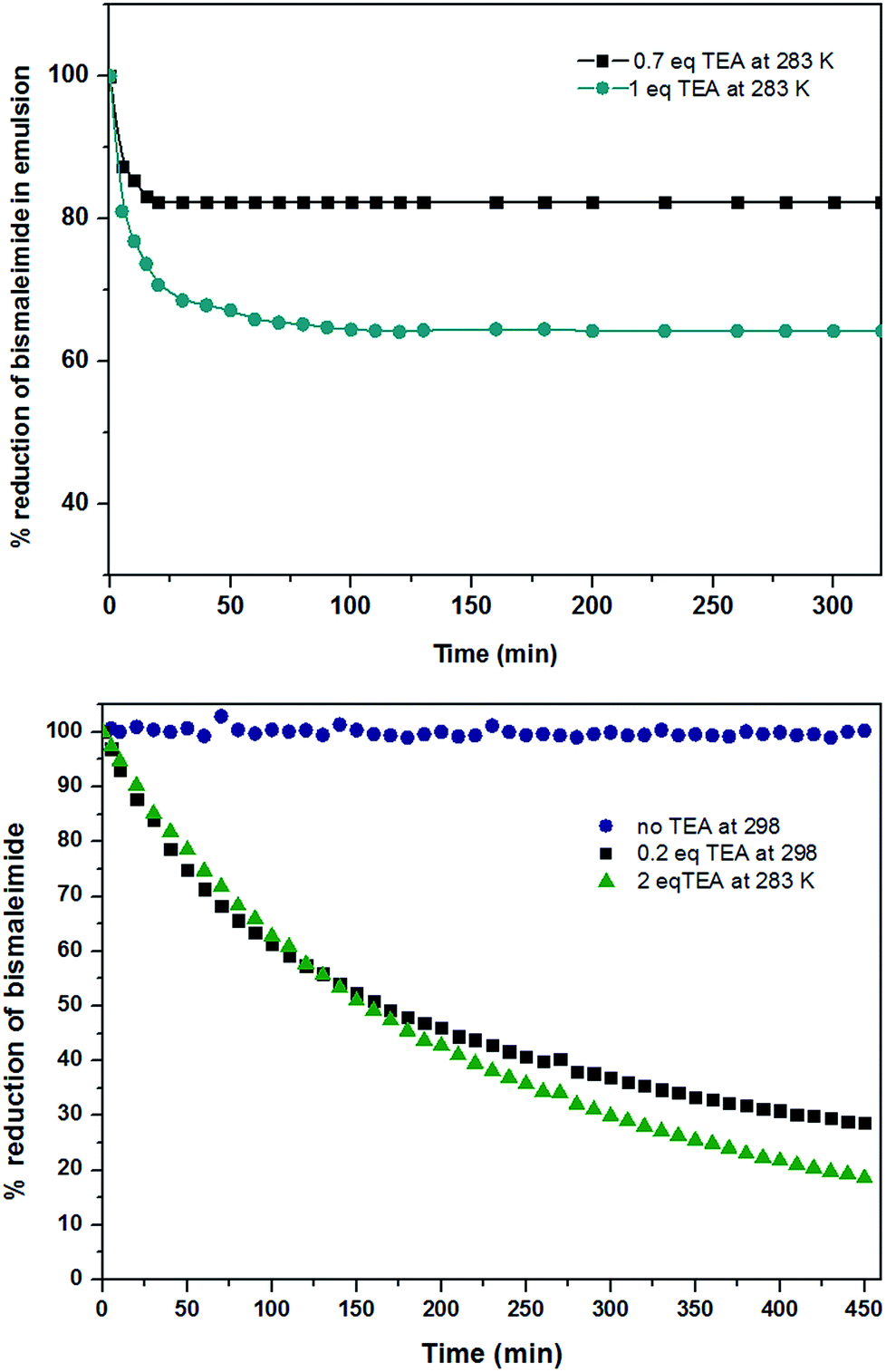 | ||
| Fig. 1 Real-time 1H NMR measurements of the thiol–maleimide reaction in solution with different amounts of TEA as catalyst, (bottom) measured in THF, [3] = [2] = 0.013 mol L−1 and in miniemulsion (top, 283 K). | ||
Both kinetic profiles prove the significant effect of TEA promoting the thiol–maleimide reaction. In contrast, the thiol–disulfide exchange reaction proceeds without the addition of the catalyst with very fast reaction kinetics in THF (at 298 K). The consumption of 1 reached more than 90% after five minutes at ambient temperature (298 K) which was too fast to measure by NMR (due to time losses for locking and shimming). However, the reduction of the temperature to 283 K and the addition of 1 eq. TEA, allowed us to bring both reaction kinetics closer to each other (Fig. 2 – still the thiol–exchange is ca. 3 times faster than that of thiol–maleimide click reaction in solution). In the miniemulsion, also for the competitive reactions a drastic increase of the reaction kinetics can be observed (0.65 mL CDCl3, 0.1 mL water and with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 eq. of TEA:monomer 3 in aqueous phase). Also very obvious is the much faster thiol–disulfide exchange leading to almost full conversion of 1, while again the slower reaction kinetics of the thiol–maleimide addition leads to incomplete consumption of 2 as the polymer membrane probably (due to the incorporation of aromatic units) hinders the reaction to reach completion. More than 70% reduction of 1 was observed in the first 10 min, after that the rate decreased, but the reaction still continues. In the case of the thiol–maleimide reaction, the rate seems to be very similar to the thiol–disulfide exchange for the first 10 min. As in contrast to solution polycondensations/-additions, the interfacial reaction setup is rather independent of monomer stoichiometry to generate polymeric material, the formation of a polymer membrane is very likely, leaving unreacted 2 and 3 (acting as cargo) behind, while the nanocapsule wall is mainly formed from the polycondensation product of 1 and 3, with several units stemming from the polyaddition product of 2 and 3 (Fig. S10† shows the TEM image of the nanocapsules obtained from this process). The molecular weight determined from GPC of the final products for the competitive “click” polyaddition/condensation is lower for the solution setup (oligomers with 1500 g mol−1) than for the miniemulsion setup (4600 g mol−1, Fig. S11†).
:1 eq. of TEA:monomer 3 in aqueous phase). Also very obvious is the much faster thiol–disulfide exchange leading to almost full conversion of 1, while again the slower reaction kinetics of the thiol–maleimide addition leads to incomplete consumption of 2 as the polymer membrane probably (due to the incorporation of aromatic units) hinders the reaction to reach completion. More than 70% reduction of 1 was observed in the first 10 min, after that the rate decreased, but the reaction still continues. In the case of the thiol–maleimide reaction, the rate seems to be very similar to the thiol–disulfide exchange for the first 10 min. As in contrast to solution polycondensations/-additions, the interfacial reaction setup is rather independent of monomer stoichiometry to generate polymeric material, the formation of a polymer membrane is very likely, leaving unreacted 2 and 3 (acting as cargo) behind, while the nanocapsule wall is mainly formed from the polycondensation product of 1 and 3, with several units stemming from the polyaddition product of 2 and 3 (Fig. S10† shows the TEM image of the nanocapsules obtained from this process). The molecular weight determined from GPC of the final products for the competitive “click” polyaddition/condensation is lower for the solution setup (oligomers with 1500 g mol−1) than for the miniemulsion setup (4600 g mol−1, Fig. S11†).
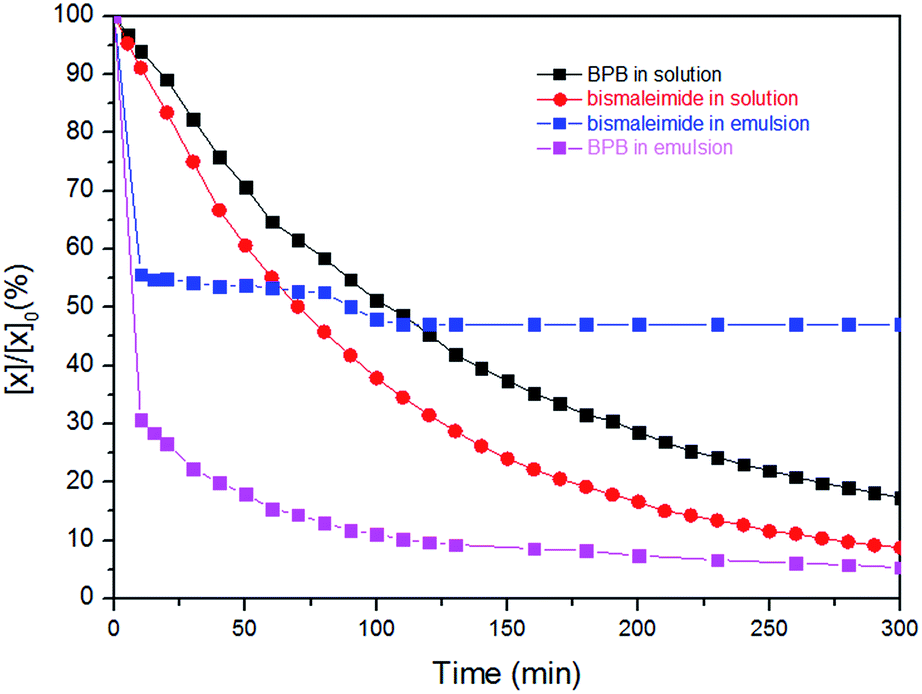 | ||
| Fig. 2 Comparison of kinetic profiles for the simultaneous reactions of thiol–disulfide interchange and thiol–maleimide in solution and miniemulsion. | ||
To determine the reaction order of the simultaneous reactions of the maleimide–thiol–disulfide reaction in solution, we followed the consumption of both bismaleimide and disulfide with 1 eq. of TEA at 283 K. It was found that the first-order reaction fitted the best in both cases. The rate constant of the thiol–disulfide exchange reaction in the competing system was 0.0072 min−1 which was a small decrease comparing with the single reaction. In contrast to the thiol–maleimide, an increase from 0.0056 min−1 to 0.014 min−1 was detected. From the 1H NMR of the final product, the contribution of monomers 1 and 2 in the final product from solution, were ca. 43% and 57%, respectively. This indicated that TEA as a basic catalyst accelerates the thiol–maleimide reaction to a higher degree than the thiol–disulfide exchange reaction. Typically, the rate of both reactions is affected by several factors i.e. pKa of thiol, nucleophilicity, solvent.19–21 However, in the thiol–disulfide reaction also the stability of the leaving group has a certain effect on the kinetics. This reason could lead to the slower rate in comparison to the thiol–maleimide reaction and then the consumption of 2 is slower than 1. From these results, the composition of the resulting polymer from the simultaneous reactions can be adjusted by the base catalyst concentration. When the base concentration (TEA) is increased; the reaction rate of the thiol–maleimide click reaction can compete with the thiol–disulfide interchange reaction resulting in high maleimide incorporation in the final product.
The results of the TOCSY and 1H NMR experiments for the product of the competing reactions were used to analyze the final product from the miniemulsion (cf. Fig. S12†). The aromatic protons from the bismaleimide are detected at 7.18 and 7.36 ppm. Also the signals of the leaving group (i.e. pyridine-2-thiol) from thiol–disulfide exchange reaction are detected at 6.75, 7.28, 7.42 and 7.65 ppm, correlating with each other. From the integration, the two other resonances at 7.78 and 7.91 ppm can be assigned to the N–H-bonds in the bucillamine and disulfide units, respectively, which slightly shift to lower magnetic field after the reaction. In addition, the 2D 1H,13C-HSQC was used to analyze the polymer obtained by miniemulsion polymerization (cf. Fig. S13†). From the H–C correlation, the signals at 7.18 and 7.36 ppm (in the 1H NMR) are related to the aromatic C–H signals of the bismaleimide at 127.5 and 130 ppm while the two signals at 7.78 and 7.91 ppm do not correlate with any carbon signals since they belong to the N–H bond.
The morphology of nanocapsules from the inverse miniemulsion process was confirmed by transmission electron microscopy (Scheme 3 and ESI†) with diameters of ranging between 300 and 500 nm as determined by dynamic light scattering after redispersing the nanocapsules in 0.1%wt aqueous SDS solution. These results confirmed the successful reactions both in solution and miniemulsion and the formation of polymeric nanocarriers by the two competing click reactions. The degradation of the nanocarriers by cleavage of the S–S-bonds via glutathione is detectable. The hydrophilic fluorescent dye sulforhodamine (SR101) was encapsulated, which is released upon degradation of the capsule shell. Fig. S14† shows higher release of SR101 for the cleavage by glutathione at room temperature after incubation over a period of 7 hours at pH 7 compared to control sample. This indicated the formation of glutathione responsive capsules via the simultaneous maleimide–thiol–disulfide reaction prepared by inverse miniemulsion.
In summary, real-time 1H NMR spectroscopy revealed a new insight into two competitive “click” reactions for the formation of polymeric nanocarriers. The competitive nucleophilic addition of a dithiol (3) with a bismaleimide (2) and the dithiol–exchange reaction with (1) was studied. The dithiol–exchange follows the faster reaction kinetics under the investigated conditions. However, the concurrent thiol–maleimide addition could be accelerated by the addition of triethylamine as a basic catalyst, allowing a copolymerization of all three monomers both in solution and at the interface of an inverse miniemulsion. The interfacial polymerization leads to higher molecular weights and exhibits faster reaction kinetics and does not need exact monomer stoichiometry. The competitive reaction at the interface produces polymeric nanocarrier with cleavable disulfide bonds. These materials may be useful drug carriers that are encapsulated in the nanocarrier or polymerized in the shell of the nanocarrier which can be released under biological conditions due to a reduction of the S–S-bonds.
Notes and references
- J. W. Chan, C. E. Hoyle and A. B. Lowe, Sequential Phosphine-catalyzed, Nucleophilic Thiol-ene/Radical-mediated Thiol-yne Reactions and the Facile Orthogonal Synthesis of Polyfunctional Materials, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, Thiol–enes: chemistry of the past with promise for the future, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, Robust, Efficient, and Orthogonal Synthesis of Dendrimers via Thiol-ene “Click” Chemistry, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS PubMed.
- L. A. Connal, C. R. Kinnane, A. N. Zelikin and F. Caruso, Stabilization and Functionalization of Polymer Multilayers and Capsules via Thiol-Ene Click Chemistry, Chem. Mater., 2009, 21, 576–578 CrossRef CAS.
- A. Gress, A. Völkel and H. Schlaad, Thio-Click Modification of Poly[2-(3-butenyl)-2-oxazoline], Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Convergent synthesis of 3-arm star polymers from RAFT-prepared poly(N,N-diethylacrylamide) via a thiol-ene click reaction, Chem. Commun., 2008, 4959–4961 RSC.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Nucleophile-Initiated Thiol-Michael Reactions: Effect of Organocatalyst, Thiol, and Ene, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- S. P. S. Koo, M. M. Stamenovic, R. A. Prasath, A. J. Inglis, F. E. Du Prez and C. Barner-Kowollik, et al., Limitations of radical thiol-ene reactions for polymer-polymer conjugation, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- J. Sun and H. Schlaad, Thiol-Ene Clickable Polypeptides, Macromolecules, 2010, 43, 4445–4448 CrossRef CAS.
- N. S. Krishnaveni, K. Surendra and K. R. Rao, Study of the Michael addition of [small beta]-cyclodextrin-thiol complexes to conjugated alkenes in water, Chem. Commun., 2005, 669–671 RSC.
- A. B. Lowe, Thiol-ene “click” reactions and recent applications in polymer and materials synthesis: a first update, Polym. Chem., 2014, 5, 4820–4870 RSC.
- K. K. Lai, R. Renneberg and W. C. Mak, Bioinspired protein microparticles fabrication by peptide mediated disulfide interchange, RSC Adv., 2014, 4, 11802–11810 RSC.
- S. Bauhuber, C. Hozsa, M. Breunig and A. Göpferich, Delivery of Nucleic Acids via Disulfide-Based Carrier Systems, Adv. Mater., 2009, 21, 3286–3306 CrossRef CAS PubMed.
- U. Paiphansiri, G. Baier, A. Kreyes, D. Yiamsawas, K. Koynov and A. Musyanovych, et al., Glutathione-Responsive DNA-Based Nanocontainers Through an “Interfacial Click” Reaction in Inverse Miniemulsion, Macromol. Chem. Phys., 2014, 215, 2457–2462 CrossRef CAS.
- B. H. Northrop, S. H. Frayne and U. Choudhary, Thiol-maleimide “click” chemistry: evaluating the influence of solvent, initiator, and thiol on the reaction mechanism, kinetics, and selectivity, Polym. Chem., 2015, 6, 3415–3430 RSC.
- A. B. Lowe, Thiol-ene “click” reactions and recent applications in polymer and materials synthesis, Polym. Chem., 2010, 1, 17–36 RSC.
- E. M. Alexandrino, P. Buchold, M. Wagner, A. Fuchs, A. Kreyes and C. K. Weiss, et al. A molecular “screw-clamp”: accelerating click reactions in miniemulsions, Chem. Commun., 2014, 50, 10495–10498 RSC.
- A. Alkan, A. Natalello, M. Wagner, H. Frey and F. R. Wurm, Ferrocene-Containing Multifunctional Polyethers: Monomer Sequence Monitoring via Quantitative 13C NMR Spectroscopy in Bulk, Macromolecules, 2014, 47, 2242–2249 CrossRef CAS.
- R. D. Bach, O. Dmitrenko and C. Thorpe, Mechanism of Thiolate-Disulfide Interchange Reactions in Biochemistry, J. Org. Chem., 2008, 73, 12–21 CrossRef CAS PubMed.
- R. Singh and G. M. Whitesides, Comparisons of rate constants for thiolate-disulfide interchange in water and in polar aprotic solvents using dynamic proton NMR line shape analysis, J. Am. Chem. Soc., 1990, 112, 1190–1197 CrossRef CAS.
- P. Nagy, Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways, Antioxid. Redox Signaling, 2013, 18, 1623–1641 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, additional spectra, and electron microscopy images, additional degradation studies. See DOI: 10.1039/c6ra08880e |
| This journal is © The Royal Society of Chemistry 2016 |