
[3,3]-Sigmatropic rearrangement mediated synthesis of chiral building blocks for the preparation of Gemini and its analogs†
Gonzalo Pazos,
Manuel Pérez,
Zoila Gándara,
Generosa Gómez* and
Yagamare Fall*
Departamento de Química Orgánica, Facultad de Química and Instituto de Investigación Biomedica (IBI), University of Vigo, Campus Marcosende, 36310 Vigo, Spain. E-mail: ggomez@uvigo.es; yagamare@uvigo.es
First published on 20th June 2016
Abstract
A novel synthetic methodology for the preparation of Gemini vitamin D3 analogs has been developed. Our procedure uses a key sigmatropic rearrangement which allows control over the C-20 stereochemistry, providing a versatile method to introduce novel side-chains to the vitamin D scaffold giving access to new analogs with potentially interesting biological properties.
Introduction
Next to its classical activities, 1α,25-dihydroxyvitamin D3 (1, calcitriol) (Fig. 1) has been shown to inhibit cellular proliferation, to induce cellular differentiation and to have numerous indirect effects on the immune system.1 However the therapeutical utility of 1 is hampered by the effective doses leading to calcemic side effects and this has stimulated the search for analogues having a relatively weak systemic effect on calcium metabolism while maintaining potent regulatory effects on cell differentiation and proliferation. Therefore, numerous synthetic vitamin D analogs have been developed for better efficacy with decreased calcemic toxicity. Among the many new calcitriol analogs, worth mentioning those in which the C-21 methyl group was extended to form a second side-chain giving rise to new class of derivatives, known as Gemini (Fig. 1). The first example of this type of compounds featured a calcitriol analog with two identical side chains and was coined Gemini.2 With the two side chains in Gemini, the possibility of chain modification is increased and the new class of Gemini analogs has significantly enlarged the biological spectrum of calcitriol.3 In spite of the obvious interest of the pharmaceutical industry and the vitamin D research groups for the Gemini analogs, the current methodologies for the preparation of these analogs lack flexibility and efficiency in the incorporation of molecular diversity. To the best of our knowledge the only known methodology to access Gemini analogs was described by Uskokovic and co-workers.2,3 | ||
| Fig. 1 Structures of 1α,25(OH)2D3, Gemini and some of its analogs. | ||
Their procedure relies on an ene reaction for the non-selective generation of the double side precursor and the subsequent separation of isomers.
Results and discussions
We anticipated that we could easily access chiral C-20 epimeric esters 5 and 6 from ketone 2 (ref. 4) through a [3,3]-sigmatropic rearrangement of allylic alcohols 3 and 4 as depicted in Scheme 1. | ||
| Scheme 1 Retrosynthetic analysis of C-20 epimeric esters 5 and 6. | ||
Accordingly, chiral ester 5 was prepared as outlined in Scheme 2.
 | ||
| Scheme 2 Synthesis of ester 5. | ||
Stereoselective Horner–Wadsworth–Emmons reaction with the anion of triethylphosphonoacetate provided the (E)-α,β-unsaturated ester 7 in 94% yield as the only stereoisomer isolated from the reaction mixture.5 Dibal-H reduction of ester 7, followed by protection of the primary alcohol gave the compound 9 in good yields. Compound 9 on reaction with selenium dioxide afforded allylic alcohol 3 in 90% yield. The ene-like SeO2 oxidation occurs exclusively on the α face of the D ring due to the steric hindrance of the β-C-18 methyl group and β-C-8 dimethyl tert-butylsiloxy group. A Johnson-orthoester Claisen rearrangement6 was performed by reaction of 3 with trimethylorthoacetate in the presence of catalytic amount of 2,4,6-trimethylbenzoic acid (TMBA) at 140 °C, affording ester 5 in 83% yield.
We thought that instead of carrying out a Johnson-orthoester Claisen rearrangement on allylic alcohol 3 we could also use a Claisen rearrangement and get another useful building block such as aldehyde 11 (Scheme 3).
 | ||
| Scheme 3 Synthesis of aldehyde 11. | ||
Reaction of allylic alcohol 3 with ethyl vinyl ether and Hg(OAc)2 afforded the desired enol ether, which after purification by column chromatography underwent a Claisen rearrangement when heated in toluene at 185 °C, giving aldehyde 11 in 90%.
Both [3,3]-sigmatropic rearrangements proceed with high stereocontrol and only one diastereomer could be detected by NMR spectroscopy.
To confirm the C-20 stereochemistry, diol 14 was prepared (Scheme 4) hoping that it might be a crystalline compound.
 | ||
| Scheme 4 Synthesis of diol 14. | ||
Catalytic hydrogenation of alkene 5 followed by Dibal-H reduction of the ester group afforded alcohol 13 in 71% overall yield. TBAF deprotection of the silyl ether group of 13 gave the diol 14 in 96% yield. To our delight, diol 14 could be recrystallized from a mixture of hexane and ethyl ether and its structure confirmed unambiguously as that shown in Fig. 2, by X-ray crystallographic analysis.7
 | ||
| Fig. 2 X-ray structure (ORTEP) of 14. | ||
For the synthesis of compound 6 we considered the same synthetic sequence using the (Z)-isomer of the allylic alcohol 8. Thus, the isomerization of the C17–C20 double bond in 8 was performed by stereoselective epoxidation of 8 [t-BuOOH, VO(acac)2] and subsequent treatment of the resulting epoxide with LiPPh2 followed by MeI.5,8 (Scheme 5). The epoxide was isolated as a single isomer and its formation occurred through the less hindered steroidal α-face.
 | ||
| Scheme 5 Isomerization of the C17–C20 double bond of 8. | ||
The (Z)-allylic alcohol 15 was then uneventfully transformed unto target compound 6 following the same synthetic sequence used for 5 (Scheme 6).
 | ||
| Scheme 6 Synthesis of ester 6. | ||
We have thus synthesized chiral building blocks 5 and 6, synthons for the synthesis of C-20 epimeric Gemini analogs.9 We now set up to demonstrate the usefulness of our method by synthesizing Gemini. Our retrosynthetic analysis for Gemini is depicted in Scheme 7.
 | ||
| Scheme 7 Retrosynthetic analysis of Gemini. | ||
We anticipated that Gemini could result from a Wittig–Horner coupling of phosphine oxide 17 and ketone 16 with the double side chain. Ketone 16 could be easily prepared from ester 5 by side chain elaboration as shown in Scheme 8.
 | ||
| Scheme 8 Synthesis of ketone 16. | ||
Catalytic hydrogenation of alkene 5 followed by Dibal-H reduction of the ester group afforded alcohol 13 in 71% overall yield. TPAP oxidation of alcohol 13 gave 90% yield of aldehyde 18 which underwent a Wittig reaction to afford alkene 19 (90%). Catalytic hydrogenation of alkene 19 gave 43% of the expected compound 20 together with alcohol 21 (48%). Compounds 20 and 21, on reaction with methyllithium afforded alcohol 22 (94%) and diol 23 (90%) respectively. Alcohol 22 reacted with TBAF to give diol 23 (94%). Iodination of the primary hydroxyl group of diol 23 gave iodide 24 in 93% yield. Nickel-mediated conjugate addition of iodide 24 to methyl acrylate10 afforded ester 25 in 92% yield. Reaction of 25 with methyllithium afforded 99% yield of diol 26. HF deprotection of the silyl ether with TBAF gave the triol 27 which upon TPAP oxidation afforded ketone 28 in 88% overall yield. TMS protection of the hydroxyl groups of 28 gave 91% yield of the target ketone 16. With ketone 16 in hand, the stage was now set for the Wittig–Horner reaction with phosphine oxide 17 (ref. 11) and the final desilylation to afford uneventfully the target Gemini compound (Scheme 9).
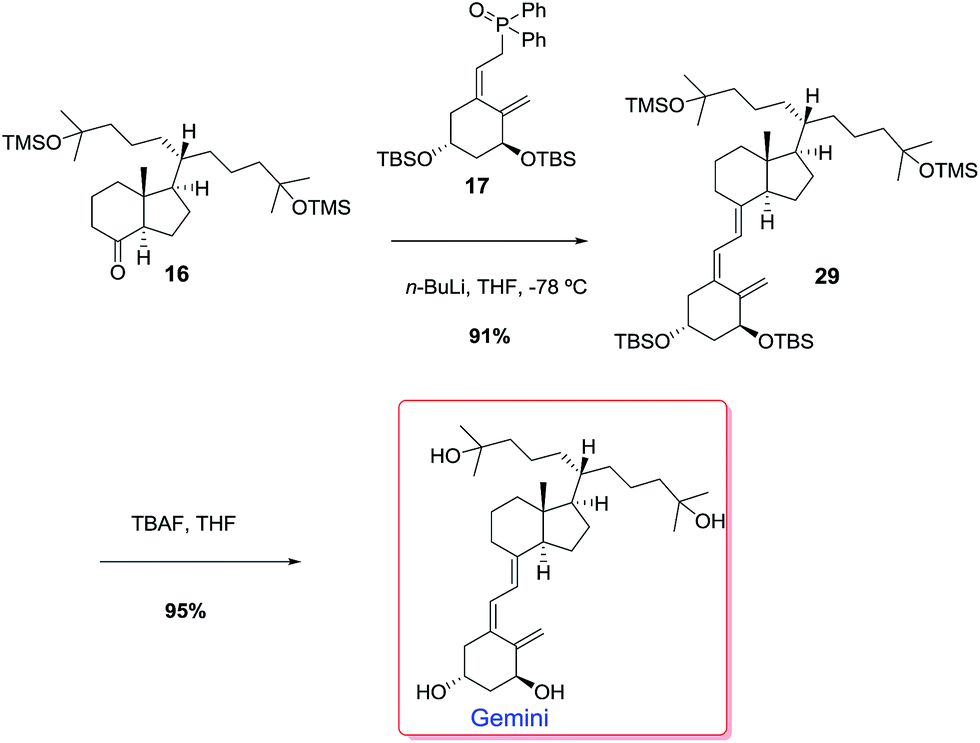 | ||
| Scheme 9 Synthesis of Gemini. | ||
Conclusion
In conclusion, we developed a new and highly flexible approach to the synthesis of chiral building blocks, useful synthons for the synthesis of Gemini analogs. Current methodologies described by Uskokovic and co-workers lack flexibility and efficiency. Our synthetic procedure uses key sigmatropic rearrangement, providing a versatile method to introduce novel side chains to the vitamin D scaffold giving access to a variety of analogs with potentially interesting biological properties. Work is now in progress for the synthesis of a series of new Gemini analogs with a view to their biological evaluation.Acknowledgements
This work was supported financially by the Xunta de Galicia (CN 2012/184). The work of the NMR and MS divisions of the research support services of the University of Vigo (CACTI) is also gratefully acknowledged. Z. G. and M. P. thank the Xunta de Galicia for Angeles Alvariño contracts.Notes and references
- (a) H. F. DeLuca, J. Burmester, H. Darwish and J. Krisinger, in Comprehensive Medicinal Chemistry, ed. C. Hansch, P. G. Sammes and J. B. Taylor, Pergamon Press, Oxford, 1991, vol. 3, pp. 1129–1143 Search PubMed; (b) H. Dai and G. H. Posner, Synthesis, 1994, 12, 1383–1398 CrossRef; (c) G.-D. Zhu and W. H. Okamura, Chem. Rev., 1995, 95, 1877–1952 CrossRef CAS; (d) G. Jones, S. A. Strugnell and H. F. DeLuca, Physiol. Rev., 1998, 78, 1193–1231 CrossRef CAS PubMed; (e) Vitamin D, ed. D. Feldman, J. W. Pike and F. H. Glorieux, Elsevier Academic Press, Burlington, MA, 2nd edn, 2005 Search PubMed; (f) Vitamin D, ed. D. Feldman, J. W. Pike and J. S. Adams, Elsevier Academic Press, San Diego, CA, 3rd edn, 2011 Search PubMed; (g) G. H. Posner and M. Kahraman, Eur. J. Org. Chem., 2003, 20, 3889–3895 CrossRef.
- A. W. Norman, P. S. Manchand, M. R. Uskokovic, W. H. Okamura, J. A. Takeuchi, J. E. Bishop, J.-I. Hisatake, H. P. Koeffler and S. Peleg, J. Med. Chem., 2000, 43, 2719–2729 CrossRef CAS PubMed.
- (a) H. J. Lee, Y. Ji, S. Paul, H. Maehr, M. Uskokovic and N. Suh, Cancer Res., 2007, 67, 11840–11847 CrossRef CAS PubMed; (b) H. J. Lee, S. Paul, N. Atalla, P. E. Thomas, X. Lin, I. Yang, B. Buckley, G. Lu, X. Zheng, Y. R. Lou, A. H. Conney, H. Maehr, L. Adorini, M. Uskokovic and N. Suh, Cancer Prev. Res., 2008, 1960, 1–9 Search PubMed; (c) J. Y. So, H. J. Lee, A. K. Smolarek, S. Paul, C.-X. Wang, H. Maehr, M. Uskokovic, X. Zheng, A. Conney, L. Cai, F. Liu and N. Suh, Mol. Pharmacol., 2011, 79, 360–367 CrossRef CAS PubMed; (d) H. Maehr and M. R. Uskokovic, Eur. J. Org. Chem., 2004, 8, 1703–1713 CrossRef; (e) P. S. Manchand and M. R. Uskokovic, US Pat., US 6008209, 1999, p. 13; (f) P. S. Manchand and M. R. Uskokovic, WO 9849138, 1998, p. 35; (g) P. S. Manchand and M. R. Uskokovic, WO 9912894, 1999, p. 60; (h) T. Huet, H. Maehr, H. J. Lee, M. R. Uskokovic, N. Suh, D. Moras and N. Rochel, Med. Chem. Commun., 2011, 2, 424–429 RSC.
- For a recent synthesis of ketone 2, see: Z. Gándara, M. L. Rivadulla, M. Pérez, G. Gómez and Y. Fall, Eur. J. Org. Chem., 2013, 5678–5682 CrossRef.
- R. Riveiros, A. Rumbo, L. A. Sarandeses and A. Mouriño, J. Org. Chem., 2007, 72, 5477–5485 CrossRef CAS PubMed.
- M. A. Hatcher and G. A. Posner, Tetrahedron Lett., 2002, 43, 5009–5012 CrossRef CAS.
- Crystallographic data were collected on a Bruker Smart 1000 CCD diffractometer at CACTI (Universidade de Vigo) at 20 °C using graphite monochromated Mo Kα radiation (λ = 0.71073 Å), and were corrected for Lorentz and polarisation effects. The frames were integrated with the Bruker SAINT software package and the data were corrected for absorption using the program SADABS. The structures were solved by direct methods using the program SHELXS97. All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2 using the program SHELXL97. Hydrogen atoms were inserted at calculated positions and constrained with isotropic thermal parameters. The structural data have been deposited with the Cambridge Crystallographic Data Centre (CCDC) with reference number CCDC 1017390.†.
- (a) E. Vedejs and P. L. Fuchs, J. Am. Chem. Soc., 1971, 93, 4070–4072 CrossRef CAS; (b) E. Vedejs and P. L. Fuchs, J. Am. Chem. Soc., 1973, 95, 822–825 CrossRef CAS.
- Y. Fall, G. Gómez, M. Pérez, Z. Gándara, X. Pérez, G. Pazos and G. Kurz, PCT Int. Appl. WO 2011121152 A1 20111006, 2011.
- S. Sinishtaj, H. B. Jeon, P. Dolan, T. W. Kensler and G. H. Posner, Bioorg. Med. Chem., 2006, 14, 6341–6348 CrossRef CAS PubMed.
- (a) A. Mouriño, M. Torneiro, C. Vitale, S. Fernández, J. Pérez-Sestelo, S. Anne and C. Gregorio, Tetrahedron Lett., 1997, 33, 4713–4716 CrossRef; (b) A. R. Daniewski, L. M. Garofalo, S. D. Hutchings, M. M. Kabat, W. Liu, M. Okabe, R. Radinov and G. P. Yiannikour, J. Org. Chem., 2002, 67, 1580–1587 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic data of all new compounds. CCDC 1017390. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra08789b |
| This journal is © The Royal Society of Chemistry 2016 |