
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic approaches to nucleopeptides containing all four nucleobases, and nucleic acid-binding studies on a mixed-sequence nucleo-oligolysine†
Giovanni N.
Roviello‡
*a and
Domenica
Musumeci‡
*ab
aCNR, Istituto di Biostrutture e Bioimmagini, Via Mezzocannone 16, 80134 Napoli, Italy. E-mail: giovanni.roviello@cnr.it; Fax: +39-81-2534574; Tel: +39-81-2534585
bUniversità di Napoli “Federico II”, Dipartimento di Scienze Chimiche, 80126 Napoli, Italy. E-mail: domenica.musumeci@unina.it; Tel: +39-81-674143
First published on 29th June 2016
Abstract
In this article we describe two solid-phase synthetic routes to obtain a nucleo-oligolysine α-peptide containing all four natural nucleobases. The first one is based on the oligomerization of the nucleobase-containing monomers, easily synthesized as herein described. The second strategy has the advantage of avoiding the solution synthesis of the monomeric building blocks, leading to the final nucleopeptide by direct solid-phase couplings of the suitably protected nucleobases with the free amino groups on the growing peptide chain still anchored to the resin. Both strategies are general and can be applied to the synthesis of nucleopeptides having backbones formed by any other diamino acid moiety decorated with the four nucleobases. We also report the CD and UV studies on the hybridization properties of the obtained nucleopeptide, containing all four nucleobases on alternate lysines in the sequence, towards complementary DNA and RNA strands. The nucleo-oligolysine with a mixed-base sequence did not prove to bind complementary DNA, but was able to recognize the complementary RNA forming a complex with a higher melting temperature than that of the corresponding RNA/RNA natural duplex and comparable with that of the analogous PNA/RNA system.
Introduction
Several artificial oligonucleotide analogues have been recently introduced in the literature, serving e.g. as antisense agents.1,2 One of the most successful in that role, PNA (peptide nucleic acids),3 is however plagued by poor water solubility and a tendency to self-aggregate. These limits can be overcome by adding L-lysine units at the PNA termini or introducing charged units (e.g. arginine) into the PNA backbone.4 This feature, along with the possibility to promote electrostatic interactions with the phosphate moieties of the nucleic acids, recently inspired us to replace the PNA backbone with an oligo-α-L-lysine backbone,5,6 in which the L-lysine units are decorated on every second residue with carboxymethyl-thymine moieties, thus leading to cationic oligomers, well soluble in water and aqueous buffers. Other examples of nucleic acid analogs based on nucleobase-bearing α-L-lysine units, similar to those employed in our works, were also described by Wada et al.7 who found them able to bind complementary RNA strands.Our previous studies on the thymine-based oligolysine suggested that base-driven interactions have a major role in the nucleic acid-binding, with respect to the expected electrostatic interactions. Furthermore, CD titrations with both homoadenine DNA and RNA sequences gave a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 base/base complex stoichiometry, on the contrary, upon replacing the undecorated lysine spacers between two adjacent thymine-functionalized residues with L-arginine residues, complexes characterised by a 2:1 T/A binding stoichiometry were observed.8
:1 base/base complex stoichiometry, on the contrary, upon replacing the undecorated lysine spacers between two adjacent thymine-functionalized residues with L-arginine residues, complexes characterised by a 2:1 T/A binding stoichiometry were observed.8
Nevertheless, these findings did not enable us to obtain detailed information on the nature of the complexes formed between the homothymine oligolysine and the homoadenine nucleic acids. To this purpose, specific hybridization experiments with mixed-sequence nucleopeptides were required to prove the ability of this oligomer to recognize complementary DNA or RNA sequences by sequence-specific hybridization, not aided by the possible formation of a ternary complex stabilized by Hoogsten-type recognition, as in the case of PNA triplexes typically formed when homopurine and homopyrimidine strands are mixed.9
In this context, we designed and realized an oligomer with an oligo-α-L-lysine backbone bearing all the four nucleobases, and here reported the CD and UV-binding experiments with complementary DNA and RNA strands. We also described two alternative synthetic approaches for the preparation of nucleobase-bearing oligo-α-L-lysines with mixed-base sequences, which could be easily extended to other nucleopeptides with the backbone based also on different amino acids. One of the synthetic routes is based on the solid-phase oligomerization of the nucleo-lysine monomers, ad hoc synthesized, coupled in alternate manner with respect to underivatized lysines. The second strategy involves the on-line assembly of both the oligolysine backbone and the suitably protected carboxymethyl-nucleobases directly on the solid support, thus avoiding some synthetic and chromatographic steps for the obtainment of the nucleoamino acid monomers.
Results and discussion
Nucleobase-amino acid conjugates, such as nucleoamino amides and nucleopeptides,10–15 are interesting molecular systems endowed with several biological properties including the ability to interact with nucleic acids. Among others, a very important biological target is poly(A), an important RNA target involved in several biological processes,16 whose ligands show high potential as therapeutics and are, thus, extensively investigated.17–20 Diamino acid-based nucleopeptides with homonucleobase sequences are easily obtainable by solid-phase peptide synthesis using protected nucleoamino acid monomers (ad hoc synthesized being generally not commercially-available),5 or by first assembling the desired peptide backbone using e.g. Dde/Fmoc protected monomers, followed by final nucleobase decoration, after selective Fmoc removal, using a molar excess of the suitable nucleobase acetic acid.21 For both strategies, properly protected amino acids can be introduced, whose side-chain groups can be regenerated after the insertion of the bases, in order to have a spacer between two consecutive nucleobase-functionalized amino acids, as well as useful moieties such as the guanidinium or ammonium groups.The synthesis of a mixed-sequence nucleopeptide is more complex than that of a homosequence oligomer requiring four different, non-commercially available monomers that have to be ad hoc synthesized.
Synthesis of the Fmoc-protected nucleolysines and their use for the nucleopeptide synthesis
In the present study we first realized the new A, C and G nucleolysines (see Fig. S1†) adopting the same synthetic strategy we previously used to obtain the thymine monomer5 (Scheme 1A). The four nucleoamino acids have been then used in the oligomerization of a mixed-sequence oligolysine by solid-phase peptide synthesis using PyBOP/DIEA as activating system (Scheme 1B).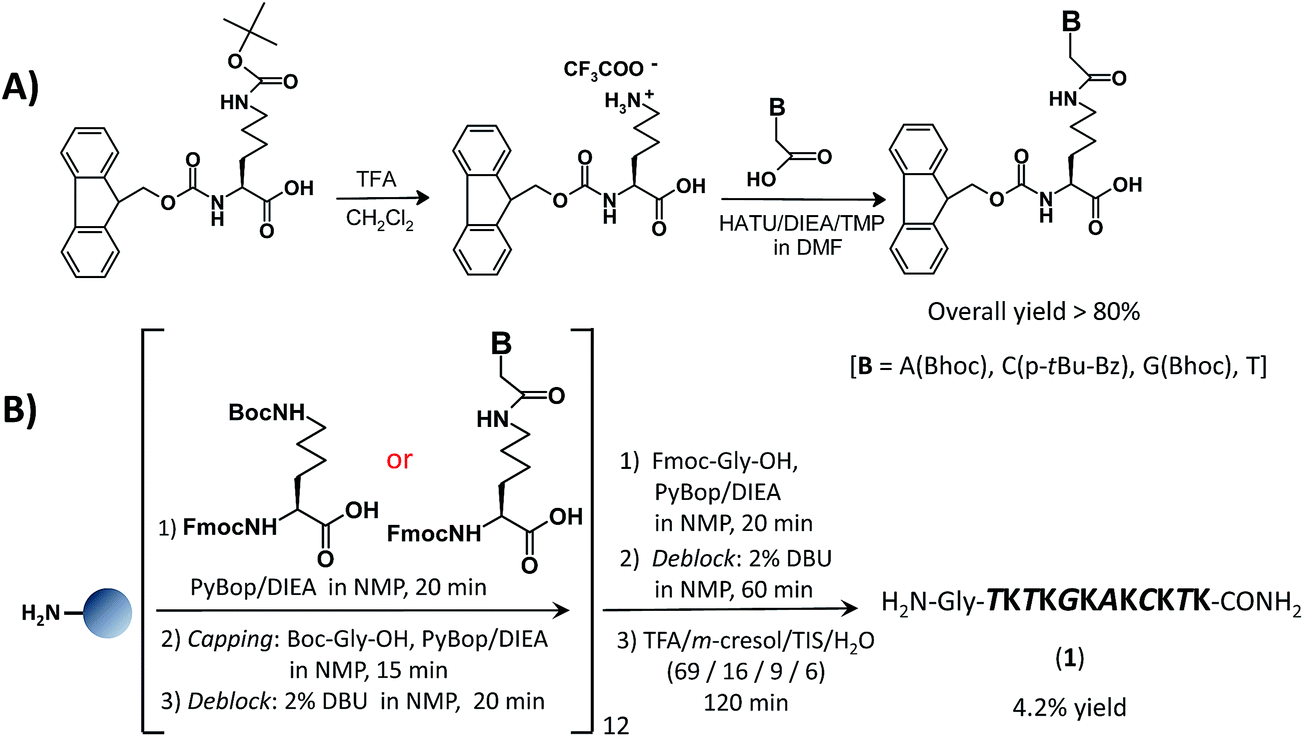 | ||
| Scheme 1 Synthetic schemes for the preparation of (A) the nucleoamino acid monomers and (B) the nucleopeptide of mixed-base sequence 1. | ||
The LC ESI-MS profiles for the nucleoamino acids and nucleopeptide 1, both well soluble in aqueous solutions, are showed in Fig. S2 (ESI†) and Fig. 1, respectively. However, the yield for this oligomerization was low (less than 5%) since the coupling of the A, C, G nucleoamino acids was more difficult than with unfunctionalized lysine and thymine nucleoaminoacid, probably due to the steric hindrance caused by the nucleobase protecting groups.
 | ||
| Fig. 1 LC-ESIMS (positive ions) of nucleopeptide NH2Gly-TKTKGKAKCKTK-CONH2 (1) (RP C18 column, HPLC method: 5 to 40% of B in A over 10 minutes). | ||
In summary, the above-described synthetic approach had two major disadvantages: (1) it required the synthesis and purification of all the four nucleolysines, and (2) afforded the desired nucleopeptide in low yield. For this reason, special efforts have been subsequently devoted to the design of a more convenient and less time-demanding strategy for the realization of the heteronucleobase-functionalised nucleopeptide (Fig. S3†). Thus, we developed a solid-phase synthetic procedure using as starting materials the carboxymethyl-nucleobases, protected with acid-labile groups on the aromatic amino groups, and two different protected lysine monomers, i.e. the commercially available Fmoc-L-Lys(Boc)-OH and Dde-L-Lys(Fmoc)-OH. The proper nucleobase sequence was obtained by an on-line solid-phase strategy. More in detail, first, the lysine monomer with the Boc semi-permanent protecting group on its side chain, was anchored onto the amino group of the Rink-amide resin (step a, Scheme 2); then, after Fmoc-removal, the coupling with the Dde-Lys(Fmoc)-OH was accomplished (step b, Scheme 2).
 | ||
| Scheme 2 Schematic representation of the on-line synthetic route to the oligolysine nucleopeptide of mixed-base sequence 2. | ||
Successively, a rapid treatment with 2% DBU in DMF allowed the selective removal of the Fmoc group from the lysine side chain in the presence of the Dde protection: the obtained free ε-amino group was then coupled with the desired carboxymethyl-nucleobase (step c, Scheme 2). Removal of Dde from the lysine α-amino group was achieved by treating the resin with a 2% hydrazine solution in DMF in order to perform the nucleopeptide-chain elongation. Steps a, b and c were repeated until the nucleopeptide with the desired base sequence was obtained. Following this protocol, a dodecapeptide oligolysine containing six nucleobases (2, Scheme 2) was achieved with a 31% overall yield, in terms of the peptide backbone (as determined by UV measurements of the released Fmoc groups). Since the efficiency of the single nucleobase insertion cannot be determined on-line during the solid-phase synthesis, two consecutive couplings were performed to assure the best yields. After deprotection and cleavage of nucleopeptide 2 from the solid support with concentrated TFA, precipitation in cold diethyl ether and lyophilisation of the redissolved crude, HPLC purification on a RP-C18 column allowed to recover about 0.9 μmol of the purified product, i.e. 10% of the initial functionalization. ESI-MS characterization confirmed the identity of the desired nucleopeptide, which was well soluble in water as expected from the initial design (see Fig. 2).
 | ||
| Fig. 2 LC-ESIMS (positive ions) of nucleopeptide 2 (RP C18 column, HPLC method: 5 to 30% of B in A over 15 min) NH2-GKCKAKTKTKTK-CONH2. | ||
The synthetic strategy reported in Scheme 2 is general and can be adapted to obtain other nucleopeptides with mixed nucleobase sequences by using diamino acids (or other amino acids as spacers) with properly protected amino groups (possibly commercially available) and the protected carboxymethyl-nucleobases. In this way, tedious purification steps relative to the synthesis of the nucleoaminoacid monomers can be avoided.
CD and UV spectroscopic studies
Subsequently, we studied by CD and UV spectroscopies the interaction of 2 with complementary DNA and RNA single strands. To this purpose, a two-chamber quartz cell (tandem cell) was used with each of the two reservoirs containing separately the nucleopeptide and the nucleic acid (AAAT[U]GC, DNA or RNA) solution at pH 7.5 and 5 °C. First, the CD profile for the two separate solutions was recorded (Fig. 3A and S4,† black lines).Successively, we recorded the CD spectrum after mixing the two solutions, (Fig. 3A and S4,† red lines). In case of DNA no appreciable change in the CD profile upon nucleopeptide addition was observed (Fig. S4A†), as it would have been expected in the case of binding with DNA. On the contrary, changes in both shape and intensity (ΔCD267 = 0.88 mdeg) of the CD spectrum with respect to the sum spectrum were observed in case of the nucleopeptide–RNA binding assay, as showed in Fig. 3A. Analogously, also sum and mix UV spectra were different in this case (Fig. 3B), and more in detail, a hypochromic effect was observed upon mixing the two ligand solutions, proving that the interaction with the nucleopeptide involved both H-bonding and stacking effects between complementary nucleobases. On the contrary, sum and mix UV-spectra in the case of the binding experiment with DNA were almost superimposable (data not shown) (Fig. 4).
 | ||
| Fig. 3 CD (A) and UV (B) profiles relative to 2 and complementary RNA solutions before (black lines) and after (red) mixing, both at a 8 μM concentration in 10 mM phosphate buffer, pH 7.5, at 5 °C (total volume after mixing = 2 ml). | ||
 | ||
| Fig. 4 UV thermal denaturation curve relative to nucleopeptide 2/RNA complex (6.7/1.5 μM respectively, in 10 mM phosphate buffer–pH 7.5; V = 3 ml). | ||
Furthermore, UV thermal denaturation experiment on the nucleopeptide–RNA complex evidenced a sigmoid-like curve with a Tm of about 26 °C consistent with the nucleopeptide–RNA binding. Remarkably, this Tm is about 10 °C higher than the corresponding melting temperature expected for r(GCAUUU)-r(AAAUGC) and d(GCATTT)-r(AAAUGC) double helices, as estimated by using Oligo-Calculator software (http://biotools.nubic.northwestern.edu/OligoCalc.html). Instead, no cooperative transition was observed in the UV-monitored melting of the system nucleopeptide/DNA (Fig. S4B, ESI†). In addition, a standard PNA of the same sequence as the nucleo-oligolysine (i.e. NH2-gcattt-CONH2) was used as reference for the stability of the RNA complex: UV-melting experiments evidenced a Tm of 28 °C for the PNA/RNA system (Fig. S5 and ESI†), which is similar to that relative to the nucleopeptide/RNA complex.
Finally, we conducted an analogous binding study with a double-stranded DNA (see Fig. S6, ESI†). Upon nucleopeptide addition, the CD spectrum of the duplex was not altered, thus showing that neither strand displacement nor formation of Hoogsteen base pairs occurred. Furthermore, UV thermal denaturation studies showed no variation of its melting temperature (data not shown).
Overall, our findings suggest that: (i) nucleo-oligolysine is able to induce conformational changes upon binding the complementary RNA strand; (ii) the structure of the formed complex is stabilized by W–C base pairs that lead to a duplex structure with stability higher than the corresponding natural counterparts (RNA/RNA and DNA/RNA) and similar to the analogous PNA/RNA system; (iii) the heteronucleobase oligolysine nucleopeptide does not alter the DNA CD bands in the here-studied conditions nor form complexes with DNA with cooperative transition upon UV-monitored melting. The latter result is apparently in contrast with our previous findings for the homothymine oligomer, in which the complex with the homoadeninic DNA was probably based on a more stable triple helix structure (involving both W–C and Hoogsteen base pairings). In any case, the ability to selectively bind RNA, discriminating complementary DNA sequences, is commonly regarded as a highly desirable feature for antisense applications. In fact, we suggest that our antisense nucleopeptide could prove beneficial in those cases in which selectivity in binding RNA and not DNA tracts is required.
Experimental section
Abbreviations
Ac2O (acetic anhydride); Bhoc (benzhydryloxycarbonyl); CD (Circular Dichroism); DBU (diazabicyclo[5.4.0]-undec-7-ene); Dde (1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)-3-ethyl); DIEA (N,N-diisopropylethylamine); DMF (dimetilformammide); Fmoc (9-fluorenylmethoxycarbonyl); HATU (O-(7-azabenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate); N-Boc Gly (N-(tert-butoxycarbonyl)glycine); NMP (4-methylpyrrolidone); NMR (Nuclear Magnetic Resonance); PyBop (benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate); TFA (trifluoroacetic acid); TIS (triisopropyl silane); TMP (2,4,6-trimethylpyridine).Chemicals
Dde-L-Lys(Fmoc)-OH, Fmoc-L-Lys(Boc)-OH, HATU, PyBOP were purchased from Novabiochem. Anhydroscan DMF and NMP were purchased from LabScan. Piperidine was Biosolve. Solvents for HPLC chromatography and acetic anhydride were purchased from Reidel-de Haën. TFA and TMP and Rink-amide resin were from Fluka. RNA and DNA oligomers in single strand form of sequence AAATGC, were purchased from Biomers. CH2Cl2, DIEA and TFA (for HPLC) were from Romil. Thyminyl acetic acid, Boc-Gly-OH, m-cresol, DBU, diethyl ether, hydrazine solution and TIS were purchased from Sigma-Aldrich. (N4-bis-Boc-cytosine-1-yl)-acetic acid was synthesised as described by Wojciechowski and Hudson.22 Bhoc-protected adenine and guanine as well as p-(tert-butyl)benzoyl-cytosine acetic acids were from ASM Research Chemicals GmbH and Co.Apparatus
Samples were analysed and characterized by LC-MS on an MSQ mass spectrometer (ThermoElectron, Milan, Italy) equipped with an ESI source operating at 3 kV needle voltage and 320 °C, and with a complete Surveyor HPLC system, comprising an MS pump, an autosampler, and a PDA detector, by using a Phenomenex Jupiter Proteo column (4 μm, 90 Å, 4.6 × 150 mm). Gradient elution was performed by building up a gradient starting with buffer A (0.05% TFA in water) and applying buffer B (0.05% TFA in acetonitrile) with a flow rate of 0.8 ml min−1 and monitoring at 260 nm.MALDI-TOF mass spectrometric analysis of the reference PNA was performed on a TOF/TOF 5800 System in positive mode, using 2,5-dihydroxybenzoic acid (DHB) as the matrix.
Analytical chromatograms were obtained on a Hewlett Packard/Agilent 1200 series HPLC, equipped with a diode array detector, by using a Phenomenex Jupiter C18 column (5 μm, 300 Å, 4.6 × 250 mm). Gradient elution was performed at 25 °C by building up a gradient starting with buffer A′ (0.1% TFA in water) and applying buffer B′ (0.1% TFA in acetonitrile) with a flow rate of 1.0 ml min−1 and monitoring at 260 nm. Semi-preparative purifications were performed on a Hewlett Packard/Agilent 1100 series HPLC, equipped with a diode array detector, by using a Phenomenex Jupiter C18 column (10 μm, 300 Å, 10 × 250 mm). Gradient elution was performed at 25 °C by building up a gradient starting with buffer A′ (0.1% TFA in water) and applying buffer B′ (0.1% TFA in acetonitrile) with a flow rate of 4.0 ml min−1 (monitoring at 260 nm). Samples were lyophilized in a FD4 Freeze Dryer (Heto Lab Equipment) for 16 h. Circular dichroism (CD) studies were conducted in analogy to other previous reports.23–25 CD spectra were obtained at 5 °C on a Jasco J-715 spectropolarimeter with a Tandem Hellma quartz cell. UV spectra and UV thermal denaturation profiles were obtained on a UV-vis Jasco model V-550 spectrophotometer equipped with a Peltier ETC-505T temperature controller, using a Hellma quartz cell with a light path of 1 cm.
Synthesis of Fmoc-protected nucleoamino acids
The synthesis of the Fmoc-protected T monomer was performed according to our previous report,5 and by analogy with this procedure we here report also the preparation of the A, C and G nucleoamino acids using for the N-ε derivatization of the lysine moiety the commercially available nucleobase acetic acids [B = A(Bhoc), T, C(p-tert-butyl benzoyl) or G(Bhoc)]. Indeed, after Boc deprotection of the Fmoc-Lys(Boc)-OH, the obtained Fmoc-protected diamino hexanoic acid (175 mg, 0.47 mmol) was dissolved in anhydrous NMP (2.0 ml), treated with DIEA (0.9 equiv., 83 μl, 0.42 mmol) and TMP (0.6 equiv., 42 μl, 0.28 mmol), and reacted with BCH2COOH (2.2 equiv., 1.03 mmol), previously preactivated by addition of HATU (2.0 equiv., 372 mg, 0.94 mmol) and DIEA (2.0 equiv., 175 μl, 0.94 mmol)/TMP (2.0 equiv., 140 μl, 0.94 mmol) in NMP (2 ml) for 2 min. After 1.5 h, the solvent was removed under vacuum and the crude was purified by RP-HPLC (RP C18 column, HPLC method: 25 to 70% of B′ in A′ over 25 min, tR ∼28 min). Yields were higher than 80% for all four monomers.Solid-phase synthesis of oligolysine nucleopeptides
800 M−1 cm−1): 84 nmol of nucleopeptide 1 (4.2% yield) were obtained; the compound was ≥95% pure by HPLC analysis. ESIMS m/z: 681.41 (found), 681.02 (expected for [C114H183N49O30 + 4H]4+); 907.41 (found), 907.69 (expected for [C114H183N49O30 + 3H]3+); 1361.07 (found), 1361.03 (expected for [C114H183N49O30 + 2H]2+).
PNA synthesis
Reference PNA (NH2-gcattt-CONH2) was obtained manually under standard conditions29–31 on a 2 μM scale. The identity of the product was checked by MALDI-TOF MS: m/z: 1648.74 (found), 1648.63 (expected for [C66H86N32O20 + H]+); 1671.68 (found), 1670.61 (expected for [C66H86N32O20 + Na]+).Conclusions
In the present work two solid-phase syntheses of cationic nucleopeptides based on L-lysine containing all the four bases were described. In one case, we first synthesized in solution all the four nucleoamino acid monomers by a simple route, and then assembled them in the solid-phase, using Fmoc-based peptide synthesis, by alternate coupling with respect to a standard Fmoc-Lys(Boc)-OH. In the second approach, in order to skip the solution synthesis of the four nucleobase-containing monomers, we developed a synthetic strategy in which the nucleobase-decoration took place on-line directly on the peptide chain growing on the solid support: in this case two kinds of differently protected lysines were coupled in alternate manner to the resin [i.e. the Fmoc-Lys(Boc)-OH and Dde-Lys(Fmoc)-OH] and the selective removal of the Dde protecting group in the presence of Fmoc was employed for the introduction of the carboxymethyl-nucleobases. The nucleopeptides obtained with both the synthetic strategies were analysed by LC-ESI MS which confirmed the identity of the expected products. These were well soluble in aqueous solutions and did not show any tendency to self-aggregate. The here described synthetic routes for the realization of the nucleo-oligolysines can be easily applied to the synthesis of other nucleopeptides with mixed base sequences starting from any other diamino carboxylic acid to be functionalised with the nucleobases, and using in combination any other natural or synthetic amino acid, possibly charged (e.g.L-arginine), as spacer unit.Furthermore, we studied the binding of the nucleo-oligolysine with mixed-base sequence towards complementary DNA and RNA strands by CD and UV spectroscopies. In the case of DNA we did not observe any significant change of the DNA CD spectrum induced by the nucleopeptide nor effects in the UV absorption spectra, i.e. the nucleopeptide did not determine any important structural rearrangement of the nucleic acid. On the other side, we found that the studied nucleopeptide bound a complementary RNA strand provoking changes in the CD spectrum of RNA. Furthermore, UV-thermal denaturation studies demonstrated that the nucleopeptide/RNA complex provided a sigmoid-like melting curve with a Tm higher than that expected in case of the corresponding RNA/RNA or DNA/RNA natural complexes. Overall, our nucleo-oligolysines seem to be promising antisense tools being able to discriminate between RNA vs. DNA complementary sequences, and forming with RNA stable complexes. Thus, all these findings encourage us to devote our future efforts on the development of molecular systems of the type described in the present study, but presenting also different nucleobase sequences and/or spacer amino acid units (e.g. having arginine in place of lysine spacers), as potential tools in biomedical strategies.
Acknowledgements
We thank Professors Antonio Roviello and Daniela Montesarchio (Federico II University, Naples – Italy) for their precious suggestions, and Mr Leopoldo Zona (IBB-CNR, Naples – Italy) for his invaluable technical assistance. We also thank Regione Campania for the research grant received for the research project “Nuovi sistemi nucleopeptidici per applicazioni diagnostiche” under the research program Legge Regionale n. 5 (annualità 2007).References
- J. H. Chan, S. Lim and W. S. Wong, Clin. Exp. Pharmacol. Physiol., 2006, 33, 533–540 CrossRef CAS PubMed.
- J. Singh, H. Kaur, A. Kaushik and S. Peer, Int. J. Pharm., 2011, 7, 294–315 CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS.
- A. Dragulescu-Andrasi, S. Rapireddy, G. He, B. Bhattacharya, J. J. Hyldig-Nielsen, G. Zon and D. H. Ly, J. Am. Chem. Soc., 2006, 128, 16104–16112 CrossRef CAS PubMed.
- G. N. Roviello, D. Musumeci, C. D'Alessandro and C. Pedone, Amino Acids, 2013, 45, 779–784 CrossRef CAS PubMed.
- G. N. Roviello, D. Musumeci, C. D'Alessandro and C. Pedone, Bioorg. Med. Chem., 2014, 22, 997–1002 CrossRef CAS PubMed.
- T. Wada, Y. Inaki and K. Takemoto, J. Bioact. Compat. Polym., 1992, 7, 25–38 CrossRef CAS.
- G. N. Roviello, C. Vicidomini, S. Di Gaetano, D. Capasso, D. Musumeci and V. Roviello, RSC Adv., 2016, 6, 14140–14148 RSC.
- P. E. Nielsen, M. Egholm and O. Buchardt, J. Mol. Recognit., 1994, 7, 165–170 CrossRef CAS PubMed.
- G. N. Roviello, V. Roviello, I. Autiero and M. Saviano, RSC Adv., 2016, 6, 27607–27613 RSC.
- G. N. Roviello, A. Ricci, E. M. Bucci and C. Pedone, Mol. BioSyst., 2011, 7, 1773–1778 RSC.
- G. N. Roviello, S. Di Gaetano, D. Capasso, S. Franco, C. Crescenzo, E. M. Bucci and C. Pedone, J. Med. Chem., 2011, 54, 2095–2101 CrossRef CAS PubMed.
- G. N. Roviello, D. Musumeci, M. Moccia, M. Castiglione, R. Sapio, M. Valente, E. M. Bucci, G. Perretta and C. Pedone, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1307–1310 CAS.
- L. Simeone, C. Irace, A. Di Pascale, D. Ciccarelli, G. D'Errico and D. Montesarchio, Eur. J. Med. Chem., 2012, 57, 429–440 CrossRef CAS PubMed.
- L. Simeone, D. Milano, L. De Napoli, C. Irace, A. Di Pascale, M. Boccalon, P. Tecilla and D. Montesarchio, Chem.–Eur. J., 2011, 17, 13854–13865 CrossRef CAS PubMed.
- M. Romano, R. van de Weerd, F. C. Brouwer, G. N. Roviello, R. Lacroix, M. Sparrius, G. van den Brink-van Stempvoort, J. J. Maaskant, A. M. van der Sar, B. J. Appelmelk, J. J. Geurtsen and R. Berisio, Structure, 2014, 22, 719–730 CrossRef CAS PubMed.
- G. N. Roviello, V. Roviello, D. Musumeci and C. Pedone, Biol. Chem., 2013, 394, 1235–1239 CrossRef CAS PubMed.
- G. N. Roviello, D. Musumeci, V. Roviello, M. Pirtskhalava, A. Egoyan and M. Mirtskhulava, Beilstein J. Nanotechnol., 2015, 6, 1338–1347 CrossRef CAS PubMed.
- G. N. Roviello, D. Musumeci and V. Roviello, Int. J. Pharm., 2015, 485, 244–248 CrossRef CAS PubMed.
- A. S. Saghyan, H. M. Simonyan, S. G. Petrosyan, A. V. Geolchanyan, G. N. Roviello, D. Musumeci and V. Roviello, Amino Acids, 2014, 46, 2325–2332 CrossRef CAS PubMed.
- G. N. Roviello, S. Di Gaetano, D. Capasso, A. Cesarani, E. M. Bucci and C. Pedone, Amino Acids, 2010, 38, 1489–1496 CrossRef CAS PubMed.
- F. Wojciechowski and R. H. Hudson, J. Org. Chem., 2008, 73, 3807–3816 CrossRef CAS PubMed.
- P. L. Scognamiglio, C. Di Natale, M. Leone, M. Poletto, L. Vitagliano, G. Tell and D. Marasco, Biochim. Biophys. Acta, 2014, 1840, 2050–2059 CrossRef CAS PubMed.
- C. Di Natale, P. L. Scognamiglio, R. Cascella, C. Cecchi, A. Russo, M. Leone, A. Penco, A. Relini, L. Federici, A. Di Matteo, F. Chiti, L. Vitagliano and D. Marasco, FASEB J., 2015, 29, 3689–3701 CrossRef CAS PubMed.
- C. Avitabile, L. Moggio, G. Malgieri, D. Capasso, S. Di Gaetano, M. Saviano, C. Pedone and A. Romanelli, PLoS One, 2012, 7, e35774, DOI:10.1371/journal.pone.0035774.
- D. Comegna, I. de Paola, M. Saviano, A. Del Gatto and L. Zaccaro, Org. Lett., 2015, 17, 640–643 CrossRef CAS PubMed.
- L. L. Vezenkov, C. A. Sanchez, V. Bellet, V. Martin, M. Maynadier, N. Bettache, V. Lisowski, J. Martinez, M. Garcia, M. Amblard and J. F. Hernandez, ChemMedChem, 2016, 11, 302–308 CrossRef CAS PubMed.
- E. Perillo, S. Porto, A. Falanga, S. Zappavigna, P. Stiuso, V. Tirino, V. Desiderio, G. Papaccio, M. Galdiero, A. Giordano, S. Galdiero and M. Caraglia, Oncotarget, 2016, 7, 4077–4092 Search PubMed.
- G. N. Roviello, D. Musumeci, E. M. Bucci, M. Castiglione, A. Cesarani, C. Pedone and G. Piccialli, Bioorg. Med. Chem. Lett., 2008, 18, 4757–4760 CrossRef CAS PubMed.
- F. Amato, R. Tomaiuolo, N. Borbone, A. Elce, J. Amato, S. D'Errico, G. De Rosa, L. Mayol, G. Piccialli, G. Oliviero and G. Castaldo, Med. Chem. Commun., 2014, 5, 68–71 RSC.
- F. Amato, R. Tomaiuolo, F. Nici, N. Borbone, A. Elce, B. Catalanotti, S. D'Errico, C. M. Morgillo, G. De Rosa, L. Mayol, G. Piccialli, G. Oliviero and G. Castaldo, BioMed Res. Int., 2014, 610718, DOI:10.1155/2014/610718.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08765e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |