
Esterification of poly(γ-glutamic acid) (γ-PGA) mediated by its tetrabutylammonium salt†
M. Biagiottia,
G. Borghesea,
P. Francescatoa,
C. F. Morellia,
A. M. Albertinib,
T. Bavaroc,
D. Ubialicd,
R. Mendichie and
G. Speranza*ad
aDipartimento di Chimica, Università degli Studi di Milano, via C. Golgi 19, I-20133 Milano, Italy. E-mail: giovanna.speranza@unimi.it
bDipartimento di Biologia e Biotecnologie “L. Spallanzani”, Università degli Studi di Pavia, via A. Ferrata 9, I-27100 Pavia, Italy
cDipartimento di Scienze del Farmaco, Università degli Studi di Pavia, viale T. Taramelli 12, I-27100 Pavia, Italy
dISTM-CNR, via C. Golgi 19, I-20133 Milano, Italy
eISMAC-CNR, via E. Bassini 15, I-20133 Milano, Italy
First published on 26th April 2016
Abstract
Poly(γ-glutamic acid) is a linear anionic biopolymer synthesized by bacterial fermentation from sustainable resources. Being water soluble, biodegradable, edible and non-toxic to humans and the environment, applications of γ-PGA are of interest in a broad range of industrial sectors. However, preparation of γ-PGA derivatives is plagued by several difficulties including its scarce solubility in organic solvents. We here report a γ-PGA derivatization procedure based on the use of its tetrabutylammonium salt. The modified solubility of γ-PGA provided by counterion exchange led to the synthesis of poly(α-ethyl-γ-glutamate), poly(α-benzyl-γ-glutamate) and poly(α-n-butyl-γ-glutamate) under smoother conditions and an almost complete functionalization degree.
Introduction
Poly(γ-glutamic acid) (γ-PGA, 1, Fig. 1) is a linear, anionic, water-soluble homopolyamide composed of D- and/or L-glutamic acid monomers polymerized via γ-amide bonds, mainly produced by Gram-positive bacteria of the genus Bacillus.1 γ-PGA is biodegradable, edible, non-toxic to humans and the environment and can be obtained by bacterial fermentation from renewable resources.1,2 The whole of these characteristics, as well as the GRAS (Generally Regarded As Safe) status of the producer bacterial strains have raised an enormous interest in γ-PGA as a novel material for many industrial and biotechnological applications.3 | ||
| Fig. 1 Poly(γ-glutamic acid) (γ-PGA, 1) and its sodium salt (2). | ||
Among others, γ-PGA has been proposed as flocculant and chelating agent in waste-water treatment, cryoprotectant and texture enhancer in food industry, moisturizer in cosmetic products, biological adhesive and drug carrier in medicine, and so on.3,4
Even more interesting perspectives arise from the possibility to chemically modify the polymer in order to modulate its chemical–physical properties and create new materials suitable for technological as well as biomedical applications.5
The synthesis of esters or amides from the free α-carboxylic groups is the most commonly used approach to attain chemically modified γ-PGA derivatives.5
However, this is a quite challenging task, given the peculiarities of γ-PGA, i.e. its scarce solubility, the high viscosity of its solutions and the inherent low chemical reactivity of the α-carboxylic side groups which are highly hindered.5 A further constraint is frequently represented by isolation and purification of the obtained products.
To date, the preparation of γ-PGA esters can be achieved by the procedure firstly reported by Kubota and coworkers.6,7 This protocol relies on the reaction of γ-PGA with alkyl halides in the presence of sodium hydrogencarbonate in an organic solvent such as DMF, DMSO or NMP at 60 °C (Scheme 1a). Under these conditions, esterification of γ-PGA with several linear alkyl halides, up to dodecyl and including dihalogenoalkanes, was carried out.5 The reaction outcome was found to be dependent on the size of the alkyl group, being better results obtained with short-chain alkyl halides.7 The derivatization step must be usually reiterated in order to achieve a high degree of functionalization. The possibility of a reduction in molecular weight upon prolonged reaction under these conditions has been also reported.8,9
 | ||
| Scheme 1 Procedures of esterification (a) and transesterification (b) of γ-PGA 5. | ||
To overcome these limitations, a two-step esterification method was developed,8 which was proved to be efficient also for the preparation of poly(γ-glutamate)s with medium or long alkyl chain (from 12 to 22 carbon atoms)10 as well as with mono-, di- and tri-ethylene glycols.11 The method consists in submitting poly(α-methyl-γ-glutamate) or poly(α-ethyl-γ-glutamate), easily obtained by the method already described, to transesterification reactions with the appropriate alcohol in the presence of Ti(OBu)4 (Scheme 1b).
However, the scarce solubility of γ-PGA in organic solvents strongly narrows the broader application of these methodologies.
Following an established strategy used in polysaccharide chemistry to prepare hyaluronic acid derivatives as well as organic solvent-soluble salts of cellulose sulfate,12,13 we here report an efficient method aimed at increasing the solubility of γ-PGA by exchanging its counterion by a quaternary ammonium salt. Such an exchange was proved to be beneficial on esterification reaction as a result of reactivity enhancement, also assisting γ-PGA manipulation.
Experimental
Materials and methods
γ-PGA Na salt (2) (Mw 28.3 kg mol−1 determined as reported below) was purchased from Natto Bioscience Co. (Japan). The acidic form (1) was prepared by treatment of an aqueous solution of 2 with 6 M HCl followed by precipitation with EtOH.All other reagents and resins were purchased from Sigma-Aldrich (Milan, Italy) and/or from VWR International (Milan, Italy) and were used without further purification. All solvents were of HPLC grade and, when necessary, they were dried on molecular sieves (3 Å). 1H NMR spectra were acquired at 400.13 MHz in D2O or in DMSO-d6 on a Bruker Advance 400 spectrometer (Bruker, Karlsruhe, Germany) interfaced with a workstation running Windows operating system and equipped with a TopSpin software package. Chemical shifts are given in ppm (δ) and are referenced to TSP [3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt, δMe 0.00 ppm] as external standard or to DMSO signal as internal standard (δH DMSO 2.50 ppm).
The functionalization degree was determined by 1H NMR analysis, in particular measuring the ratio of intensities of the backbone α-CH signal at about 4.2 ppm and of the side-chain OCH2 signal in the interval 4.04–5.1 ppm.
Molecular weight distribution (MWD)
The characterization of the molecular weight distribution (MWD) of the samples was performed by using a multi-angle laser light scattering (MALS) absolute detector on-line with a size exclusion chromatographic (SEC or GPC) system. The MWD, relative averages and dispersity indexes were obtained by a modular multi-detector SEC system. The SEC system consisted of an Alliance 2695 separation module from Waters (Milford, MA, USA) equipped with two on-line detectors: a MALS Dawn DSP-F photometer from Wyatt (Santa Barbara, CA, USA) and a 2414 differential refractometer (DRI) from Waters used as concentration detector. The on-line MALS detector provides absolute values of the molecular weight of the polymeric samples without external calibration, as previously reported.16,17The SEC-MALS experimental conditions were selected on the basis of samples solubility. γ-PGA Na salt (2) and acid form (1): columns, 2 ultrahydrogel (2000–500 Å) from Waters; mobile phase, 0.1 M NaCl + 0.1 M phosphate buffer pH 7.0; temperature, 35 °C; flow rate, 0.8 mL min−1; injection volume, 150 μL; sample concentration, about 1 or 5 mg mL−1.
γ-PGA TBA salt (3): columns, 2 TSKGel (G4000–G3000) from Tosoh; mobile phase, 0.1 M NaCl + 0.1 M phosphate buffer pH 7.4; temperature, 35 °C; flow rate, 0.8 mL min−1; injection volume, 100 μL; sample concentration, about 2 mg mL−1. γ-PGA esters (4–6): columns, 2 PLgel Mixed C from Polymer Laboratories; mobile phase, DMF + 0.05 M LiBr; temperature, 50 °C; flow rate, 0.8 mL min−1; injection volume, 100 μL; sample concentration, about 4 mg mL−1.
Preparation of γ-PGA tetrabutylammonium salt (3, γ-PGA TBA salt)
γ-PGA sodium salt (2, 1.0 g, 6.6 mmol) was dissolved in 40 mL of water and then percolated through a column filled with 10 mL of wet Dowex 50WX8-200 ion-exchange resin (capacity 1.7 meq. mL−1 by wetted bed volume), previously activated with a 1.5 M tetrabutylammonium hydroxide solution. The resin was subsequently washed 8 times with water. The eluate was collected and lyophilized to give 1.8 g (73% yield) of γ-PGA tetrabutylammonium salt (3). Ammonium ion exchange degree was >99%; Mw 31.6 kg mol−1.1H NMR (400 MHz, DMSO-d6) δ 0.94 (t, J = 7.2 Hz, 12H, –NCH2CH2CH2CH3), 1.27–1.37 (m, 8H, –NCH2CH2CH2CH3), 1.55–1.62 (m, 8H, –NCH2CH2CH2CH3), 1.67–1.82 (broad m, 2H, –CH2CH2CO–), 1.86–1.97 (broad m, 1H, –CH2CH2CO–), 1.99–2.10 (broad m, 1H, –CH2CH2CO–), 3.18–3.24 (m, 8H, –NCH2CH2CH2CH3), 3.55–3.63 (broad m, 1H, –CHCOO), 6.99 (broad d, J = 4.8, 1H, –CONH).
13C NMR (100 MHz, DMSO-d6) δ 13.93 N(CH2CH2CH2CH3), 19.67 (N(CH2CH2CH2CH3)4), 23.55 (N(CH2CH2CH2CH3)4), 28.13 (CHCH2CH2CO), 32.42 (CHCH2CH2CO), 52.85 (CHCH2CH2CO), 58.07 (N(CH2CH2CH2CH3)4), 172.07 (CONH); 174.33 (COO).
Preparation of poly(α-ethyl γ-glutamate) (4)
A higher functionalization degree (72%) was achieved by reacting this material under the same conditions for further 2 days. 270 mg of 4 (26% yield, Mw 33.4 kg mol−1) were obtained. NMR data: in agreement with those previously reported.7
Once the additions were completed the solution was stirred at r.t. for 36 hours. The reaction mixture was diluted with EtOH (10 mL) and neutralized with a saturated NaHCO3 solution (15 mL). After removal of EtOH under reduced pressure, the residue was worked-up as above to give 30 mg of 4 as a white powder (70% yield, functionalization degree: 100%, Mw 21.8 kg mol−1). NMR data: in agreement with those previously reported.7
Preparation of poly(α-benzyl γ-glutamate) (5) (procedure B2)
γ-PGA tetrabutylammonium salt (3, 100 mg, 0.27 mmol) was dissolved in 4 mL of anhydrous benzyl alcohol and the reaction was carried out as described above. The product (5, 38 mg) was obtained as a white powder. Yield: 63%; functionalization degree: 100%; Mw 39.5 kg mol−1; NMR data: in agreement with those previously reported.6Preparation of poly(α-n-butyl γ-glutamate) (6) (procedure B2)
γ-PGA tetrabutylammonium salt (3, 100 mg, 0.27 mmol) was dissolved in 12 mL of anhydrous n-butanol and the reaction was carried out as described above. The product (6, 23 mg) was obtained as a white powder. Yield: 45%; functionalization degree: 99%; Mw 20.8 kg mol−1; NMR data: in agreement with those previously reported.7Solubility assays
Each test was performed by weighting an amount of about 15 μmol of substance (corresponding to 2.0 mg, 2.3 mg and 5 mg of 1, 2 and 3, respectively) and adding stepwise 50 μL of the selected solvent till complete dissolution or up to a final volume of 2 mL. Assays were performed under magnetic stirring at room temperature for 8 hours.Results and discussion
It is well known that the nature of the counterion has an important influence on the physicochemical properties of ionic materials, including solubility. Specifically the solubility in organic solvents increases markedly when a large “onium” cation is substituted for the usual alkali metal cation.14 We reasoned therefore that the substitution of a metal counterion with a tetrabutylammonium ion would have been beneficial for the solubility profile of γ-PGA. In order to test our hypothesis, commercially available sodium salt of γ-PGA (2) was solubilized in water and the resulting solution was percolated through a column filled with the sulphonic resin Dowex 50Wx8 in the form of tetrabutylammonium salt (TBA). The eluate was collected, freeze-dried to give γ-PGA TBA salt (3) in 73% yield (Scheme 2).12 Analysis of NMR spectra indicated that under these conditions, almost a complete substitution of sodium with TBA ion occurred. | ||
| Scheme 2 Synthesis of γ-PGA TBA salt (3). | ||
Solubility tests carried out on 3 as well as on γ-PGA (1) and γ-PGA sodium salt (2) (see Table 1) indicated that the presence of the TBA counterion improves the solubility of the biopolymer in organic solvent, particularly in the case of aprotic polar solvents such as DMF and DMSO, and short-chain alcohols (MeOH, EtOH, n-PrOH). However, γ-PGA TBA salt (3) still remains insoluble in ethers and branched alcohols (iso-PrOH and t-BuOH).
| Solvent | γ-PGA (1) | γ-PGA Na (2) | γ-PGA TBA (3) |
|---|---|---|---|
| a Each test was performed by weighting an amount of about 15 μmol of substance (corresponding to 2.0 mg, 2.3 mg and 5 mg of 1, 2 and 3, respectively) and adding stepwise 50 μL of the selected solvent till complete dissolution or up to a final volume of 2 mL, under magnetic stirring at room temperature for 8 hours. Legend: −, insoluble; +, soluble; ++, very soluble; ±, sparingly soluble. | |||
| MeOH | − | − | + |
| EtOH | − | − | + |
| n-PrOH | − | − | + |
| i-PrOH | − | − | − |
| n-BuOH | − | − | ± |
| t-BuOH | − | − | − |
| Benzyl alcohol | − | − | + |
| 2,2,2-Trifluoroethanol | − | − | + |
| Diethyl ether | − | − | − |
| Dioxane | − | − | − |
| Acetonitrile | − | − | − |
| DMF | − | ± | ++ |
| DMSO | ± | ± | ++ |
To evaluate the effect of the solubility enhancement on reactivity, we investigated the esterification of the biopolymer and, specifically, the preparation of poly(α-ethyl γ-glutamate) (4) as a reference reaction. Poly(α-alkyl-γ-glutamate)s are among the most promising derivatives of PGA e.g. alkyl esters of PGA display improved thermal and mechanical properties with respect to the polyacid and have been investigated for their capability to form biodegradable fibers and films that can replace currently used non-biodegradable polymers.2,5
Two methodologies were used. The former method (namely, A) is the one already described above, firstly reported by Kubota5–7 and based on carboxylate chemistry, which was found to be effective when γ-PGA sodium salt (2) is used as the starting material (see Scheme 3, Method A).
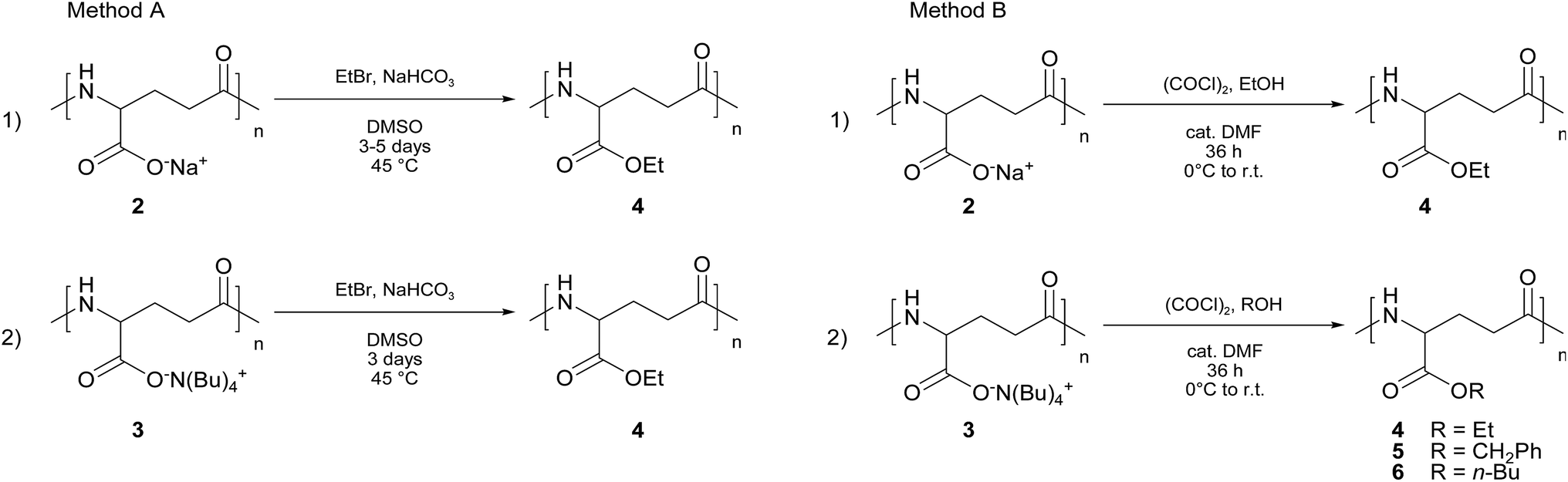 | ||
| Scheme 3 Alkylation of γ-PGA salts with ethyl bromide (Method A) and esterification with oxalyl chloride-catalytic DMF and alcohol (Method B). | ||
The latter route (namely, B) was set up in our laboratories and consists in the in situ formation of the acyl chloride of poly(γ-glutamic acid) by treatment of both 2 and 3 with oxalyl chloride in the presence of a catalytic amount of dimethylformamide (DMF),15 followed by direct esterification with the proper alcohol (see Scheme 3, Method B).
Using γ-PGA sodium salt (2) as the starting material our strategy appears advantageous with respect to the Kubota's methodology in terms of reaction time (120 hours vs. 36 hours) and final yield (26% vs. 80%). Nevertheless, the functionalization degree is far to be excellent (72% vs. 22%, see Table 2, entries A1 and B1).
| Methoda | Producta | T (°C) | Time (h) | Yield (%) | Functionalization degreeb (%) |
|---|---|---|---|---|---|
| a See Scheme 3.b The functionalization degree was determined by 1H NMR analysis, in particular measuring the ratio of intensities of the backbone α-CH signal at about 4.2 ppm and of the side-chain OCH2 signal in the interval 4.04–5.1 ppm.c Recovery due to the presence of solvent residues. | |||||
| A1 | 4 | 45 | 120 | 26 | 72 |
| A2 | 4 | 45 | 72 | 99 | 100 |
| B1 | 4 | r.t | 36 | 80 | 22 |
| B2 | 4 | r.t. | 36 | 70 | 100 |
| 5 | r.t. | 36 | 63c | 100 | |
| 6 | r.t. | 36 | 45 | 99 | |
Contrariwise, when γ-PGA TBA salt (3) was used as the starting material in both procedures, an overall improvement of the reaction outcome was observed in terms of reaction time as well as yield and functionalization degree. In the case of Method A, the reaction time was reduced from 120 to 72 hours and, mostly important, both the functionalization degree and the yield were almost complete (cf. entries A1 and A2 in Table 2).
On the other hand, the use of γ-PGA TBA salt (3) in the procedure B, under the same experimental conditions (r.t., 36 hours), afforded an almost complete functionalized product (4), although with a slightly lower yield (cf. Table 2, entries B1 and B2). It is also worth noting that Method B does not require any heating.
Since the acyl chloride-based approach gave promising results for the preparation of poly(α-ethyl γ-glutamate) (4), we extended our methodology to the synthesis of other poly(α-alkyl γ-glutamate)s, in particular benzyl and n-butyl derivatives (5 and 6, respectively). These esters were chosen taking into account the different solubility of the γ-PGA TBA (3) in benzyl and n-butyl alcohols (Table 1). For both 5 and 6 a very high functionalization degree was achieved even if accompanied by a moderate yield in the case of 6. The effect of the lower solubility of 3 in n-butyl alcohol cannot be ruled out.
Conclusions
In conclusion, γ-PGA TBA (3) was shown to be a valid starting material for a straightforward chemical modification of the biopolymer. The routinely used esterification procedure based on the reaction with alkyl bromides and the newly developed method based on the activation of the carboxylic groups with oxalyl chloride were successfully applied for the preparation of three esters (4–6). Taking into account the interest in γ-PGA esters as materials endowed with improved thermal and mechanical properties, the preparation of γ-PGA TBA and the set-up of a new method of esterification may assist the practical applications of this unexploited biopolymer.Acknowledgements
This work was financially supported by Alma Mater Ticinensis Foundation (Pavia, Italy) to A. M. A., Call 2010–2011 “Promuovere la ricerca d'eccellenza”. Project: “Poly(γ-glutamate) (γ-PGA): a biocompatible and biodegradable material for the immobilization of biologically active molecules”.Notes and references
- A. Ogunleye, A. Bhat, V. U. Irorere, D. Hill, C. Williams and I. Radecka, Microbiology, 2015, 161, 1–17 CrossRef CAS PubMed
.
- I.-L. Shih and J.-Y. Wu, in Microbial Production of Biopolymers and Polymer Precursors: Applications and Perspectives, ed. B. H. A. Rehm, Caister Academic Press, Norfolk, UK, 2009 Search PubMed
- H. Poo, C. Park, M.-S. Kwak, D.-Y. Choi, S.-P. Hong, I.-H. Lee, Y. T. Lim, Y. K. Choi, S.-R. Bae, H. Uyama, C.-J. Kim and M.-H. Sung, Chem. Biodiversity, 2010, 7, 1555–1562 CAS
- I. Bajaj and R. Singhal, Bioresour. Technol., 2011, 102, 5551–5560 CrossRef CAS PubMed
- S. Muñoz-Guerra, M. García-Alvarez and J.-A. Portilla-Arias, J. Renewable Mater., 2013, 1, 42–60 CrossRef
- H. Kubota, Y. Nambu and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2877–2878 CrossRef CAS
- H. Kubota, Y. Nambu and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 85–88 CrossRef CAS
- J. Melis, M. Morillo, A. Martínez de Ilarduya and S. Muñoz-Guerra, Polymer, 2001, 42, 9319–9327 CrossRef CAS
- A. Martínez de Ilarduya, N. Ittobane, M. Bermúdez, A. Alla, M. El Idrissi and S. Muñoz-Guerra, Biomacromolecules, 2002, 3, 1078–1086 CrossRef
- M. Morillo, A. Martínez de Ilarduya and S. Muñoz-Guerra, Macromolecules, 2001, 34, 7868–7875 CrossRef CAS
- G. Pérez-Camero, B. Vázquez and S. Muñoz-Guerra, J. Appl. Polym. Sci., 2001, 82, 2027–2036 CrossRef
- D. Bellini and A. Topai, WO2000001733 A1, 2011
- R. G. Schweiger, US 3,637,520, 1972
- E. V. Dehmlow and S. S. Dehmlow, Phase Transfer Catalysis, VCH, Weinheim, 3rd edn, 1993 Search PubMed
- A. Wissner and C. V. Grudzinskas, J. Org. Chem., 1978, 43, 3972–3974 CrossRef CAS
- R. Mendichi and A. Giacometti Schieroni, Current Trends in Polymer Science, ed. S. G. Pandalai, Trans-World Research Network, Trivandrum, India, 2001, vol. 6, pp. 17–32 Search PubMed
- P. J. Wyatt, Anal. Chim. Acta, 1993, 272, 1–40 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Molecular weight distribution (MWD) of γ-PGA (1) and γ-PGA derivatives (2–6), 1H and 13C NMR of γ-PGA TBA salt (3), 1H NMR data of poly(α-ethyl γ-glutamate) (4), poly(α-benzyl γ-glutamate) (5) and poly(α-n-butyl γ-glutamate) (6). See DOI: 10.1039/c6ra08567a |
| This journal is © The Royal Society of Chemistry 2016 |