
Flavans with cytotoxic activity from the stem and root bark of Daphne giraldii†
Abstract
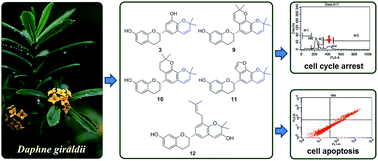
Thirteen new flavan compounds named daphnegiravans A–M (1–13) and eight known analogues (14–21) were isolated from the stem and root bark of Daphne giraldii. Their structures were established by comprehensive analysis of NMR data and CD spectra. Five human cancer cell lines (MCF-7, Bcap37, HepG2, Hep3B and A549) were used to evaluate the antitumor activity of all the isolates and their structure–activity relationships were also discussed. Interestingly, prenylated and some methoxy flavans exhibited the highest activities against Hep3B compared with the other cell lines, especially 3 and 9–12 with IC50 values ranging from 5.15 to 9.66 μM. Furthermore, flow cytometry analysis indicated that 3 and 9–11 possessing a 2,2-dimethylpyran moiety in ring B induced G2/M phase arrest in Hep3B cells, while 12 with different structural features effectively inhibited cell proliferation by evoking apoptotic cell death. The reactive oxygen species (ROS) generation might be responsible for the induction of arrest and apoptosis.
 Please wait while we load your content...
Please wait while we load your content...

