
Amphiphilic hyperbranched polymers with a biodegradable hyperbranched poly(ε-caprolactone) core prepared from homologous AB2 macromonomer†
Xiaojin Zhang*a,
Juan Chengb and
Renxi Zhuoa
aKey Laboratory of Biomedical Polymers of Ministry of Education, Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: zhangxj@whu.edu.cn
bKey Laboratory of Catalysis and Materials Science of the State Ethnic Affairs Commission & Ministry of Education, South-Central University for Nationalities, Wuhan 430074, China
First published on 25th May 2016
Abstract
Amphiphilic hyperbranched polymers with a biodegradable hyperbranched poly(ε-caprolactone) core were synthesized by one-pot polyesterification of AB2 macromonomer (including two carboxyl groups and one hydroxyl group) and poly(ethylene glycol) methyl ether, as a very promising biodegradable and biocompatible biomaterial for drug controlled release.
Amphiphilic polymers are able to form self-assembled nanostructures in a selected solvent.1 The nanostructures include micelles,2 vesicles,3 nanoparticles,4 nanospheres,5 and multicompartmental microcapsules.6 They possess some interesting shapes, such as spherical,7 cylindrical,8 rod,9 hexagonal,10 onion-like,11 and worm-like.12 Because of their special shapes in a selected solvent and their potential applications in the fields of biomedicine and nanotechnology, amphiphilic polymers have received much attention and extensive research.13 Among them, polymeric micelles with small particle size (generally less than 100 nm) are one of the most valuable drug carriers developed in recent years and have some significant advantages, such as stable structure, excellent tissue penetration, wide drug-loading application, long in vivo detention time, and targeted delivery of the drug.14
Polymeric micelles are typically prepared by self-assembly of block polymers.15 As the most common type of amphiphilic polymers, block polymers contain diblock polymers, triblock polymers, and multiblock polymers. In addition, amphiphilic polymers also exhibit the different structures, such as linear,16 comb-like,17 toothbrush-like,18 miktoarm,19 dendritic,20 Y-shaped,21 H-shaped,22 and tadpole-shaped.23 The structures of amphiphilic polymers have a great effect on the performance of polymeric micelles.24 Compared to linear block polymers, diversified structures improve the stability of polymeric micelles and enhance the drug release property of polymeric micelles.25 In the previous studies, we found that the micelles of miktoarm,26 block–graft,27,28 and comb-like29 polymers have more stable and sustained in vitro drug release than that of linear block polymers. However, amphiphilic hyperbranched polymers are rarely reported so far to form polymeric micelles. It is probably that amphiphilic hyperbranched polymers are difficult to be synthesized30 or the conventional hyperbranched polymers are non-biodegradable polymers such as polystyrene derivatives and polyether derivatives,31 which greatly limits in vivo applications of polymeric micelles.
In the past decades, only a few literature examples report on the synthesis of biodegradable hyperbranched polymers.32 For example, 4-(2-hydroxyethyl)-ε-caprolactone,33 6-hydroxymethyl-1,4-dioxan-2-one,34 5-hydroxymethyl-1,4-dioxan-2-one,35 5-{3-[(2-hydroxyethyl)thio]propoxy}-1,3-dioxan-2-one,36 and 5-(4-hydroxybutyl)-1,3-dioxan-2-one37 were synthesized and applied for the preparation of biodegradable hyperbranched polymers by self-condensing ring-opening polymerization (SCROP). However, the synthesis of these monomers requires a lot of steps and the monomers needs to be purified to high purity in order to be used for SCROP. Inspired by the smart works of Hedrick et al. and others for the preparation of hyperbranched poly(ε-caprolactone) derived from intrinsically branched AB2 macromonomers,38–41 here we design homologous AB2 macromonomer (carboxyl-terminated poly(ε-caprolactone), CPCL) with two carboxyl groups and one hydroxyl group, prepared by ring-opening polymerization of ε-caprolactone with dibenzyl malate42 as an initiator and then removing benzyl groups. Then, homologous AB2 macromonomer CPCL was used to synthesize amphiphilic hyperbranched polymers mPEG–HPCL with biodegradable hyperbranched poly(ε-caprolactone) core by one-pot polyesterification of CPCL and poly(ethylene glycol)methyl ether (mPEG) (Scheme 1). Amphiphilic hyperbranched polymers mPEG–HPCL were used to prepare polymeric micelles. The size, morphology, drug loading property, in vitro drug release behavior, and in vitro cytotoxicity of the micelles were investigated.
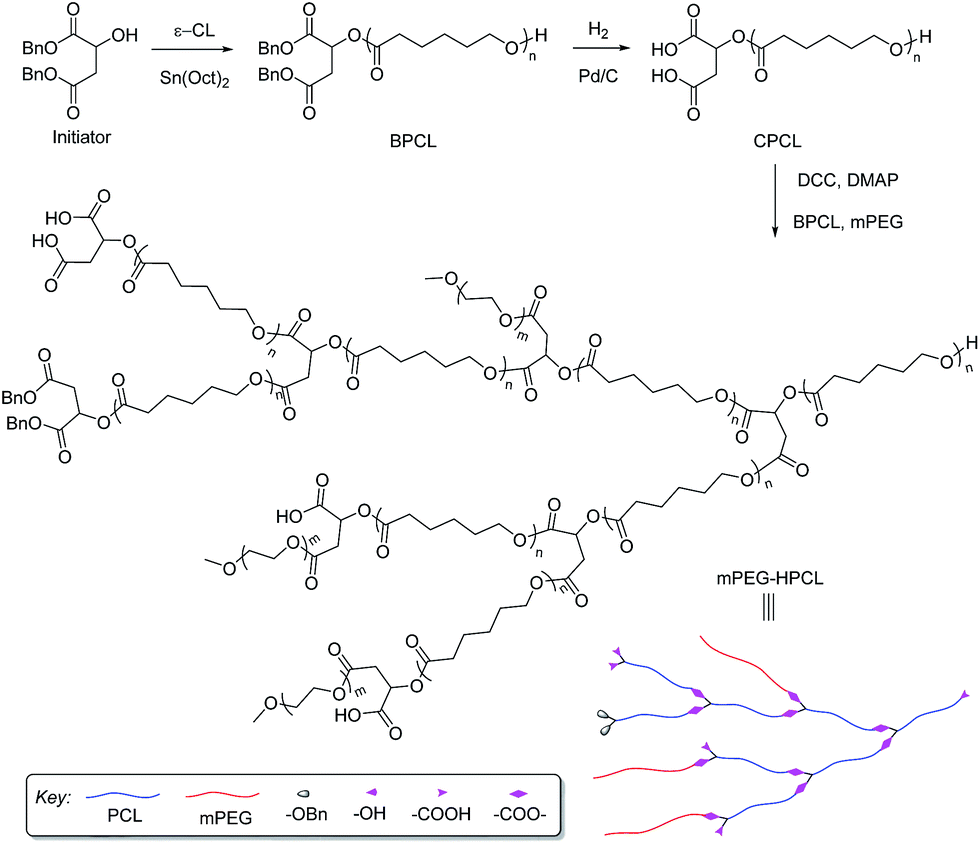 | ||
| Scheme 1 Synthesis of amphiphilic hyperbranched polymers mPEG–HPCL. | ||
In order to be able to calculate molar ratio and molecular weight of mPEG–HPCL from the 1H NMR spectrum, a small amount of benzyl-protected carboxyl-terminated poly(ε-caprolactone) (BPCL) is added and the integration of the peak areas for the benzyl groups is used as the base. We synthesized two different molecular weights of BPCL by adjusting the feed ratios of dibenzyl malate (Fig. 1a) and ε-CL in the polymerization. The hydrogenolytic deprotection of BPCL was carried out to synthesize two different molecular weights of CPCL. The synthetic results are displayed in Table S1.† The yields of BPCL and CPCL are both high. The degree of polymerization (DP) of BPCL is calculated by comparing the integrations of the signals at 5.17–5.10 ppm (PhCH2–) for the initiator group and 2.43–2.22 ppm (–CH2CH2CH2CH2CH2OCO–) for PCL segments (Fig. 1b). The molar ratios of Bn, and ε-CL in BPCL calculated from the 1H NMR spectra are close to the ratios in feed. The molecular weights were also determined by GPC. The values are close to those calculated from the 1H NMR spectra. The results indicate that BPCL are prepared in a high yield through adjusting the feed ratio of initiator and monomer. Compared to the 1H NMR spectrum of BPCL, the characteristic peaks (7.35 ppm, 5.15 ppm, and 5.11 ppm) of benzyl in the 1H NMR spectrum of CPCL in Fig. 1c disappear completely, indicating that the protecting groups of BPCL are removed completely and the debenzylation ratio is nearly 100%. The characteristic signals of terminated –CH2OH are observed from the 1H NMR spectra of BPCL and CPCL. We synthesized four amphiphilic hyperbranched polymers by adjusting the feed ratios of BPCL, mPEG, and CPCL in the polyesterification. The synthetic results are shown in Table 1. The 1H NMR spectrum of mPEG2.7–(HPCL10)5.2 (where the numbers on the lower right corner of mPEG, HPCL, and brackets stand for the grafted number of mPEG chain, the DP of ε-caprolactone in BPCL, and the conjugated number of CPCL, respectively) is shown in Fig. 1d. The characteristic peaks of benzyl group at 7.35, 5.15, 5.11, and 2.97 ppm, mPEG at 3.65 and 3.38 ppm, and PCL segments at 2.43–2.22 ppm are observed. The DPs are calculated by comparing the integration of these peaks. The Mn data calculated from 1H NMR spectra are in good agreement with the predicted Mn.
 | ||
| Fig. 1 1H NMR spectra (A) of dibenzyl malate (a), BPCL10 (b), CPCL10 (c), and mPEG2.7–(HPCL10)5.2 (d) (300 MHz, CDCl3). The characteristic signals of dibenzyl malate, mEPG, and PCL are labeled using Arabic numerals, “+”, and “×”, respectively. 1H NMR spectra in the boxes (B–D) are the partial enlarged drawings of 1H NMR spectra of BPCL10 (b), CPCL10 (c), and mPEG2.7–(HPCL10)5.2 (d). The residual solvent signal (CHCl3) is labeled using “*”. The characteristic signals of terminated benzyl groups and terminated –CH2OH are labeled using “#” and “&”. | ||
| Polymers | Feed ratioc | 1H NMR | GPC | WmPEGe | Drug-free micellef | Drug-loaded micelleg | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ratiod | Mn | Mn | PDI | Size (nm) | PDI | Size (nm) | PDI | DLCh (%) | EEi (%) | |||
a Polymerization conditions: [monomer]/[Sn(Oct)2] = 1000, 130 °C, 24 h, in bulk mPEG1 with molecular weight of 5000 Da.b DCC, DMAP, THF, room temperature, 24 h, mPEG with molecular weight of 2000 Da.c [BPCL]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[mPEG]:[CPCL] in feed.d [mPEG]:[CL] or [BnO–]:[mPEG]:[CPCL] calculated from 1H NMR spectra.e Weight fraction of mPEG chain in the polymers.f Measured at a concentration of 0.5 mg mL−1.g Measured with 10 mg of polymers and 1.0 mg of methotrexate as a model drug.h Drug loading content (DLC).i Entrapment efficiency (EE). :[mPEG]:[CPCL] in feed.d [mPEG]:[CL] or [BnO–]:[mPEG]:[CPCL] calculated from 1H NMR spectra.e Weight fraction of mPEG chain in the polymers.f Measured at a concentration of 0.5 mg mL−1.g Measured with 10 mg of polymers and 1.0 mg of methotrexate as a model drug.h Drug loading content (DLC).i Entrapment efficiency (EE). |
||||||||||||
| mPEG1-b-PCL48a | — | 1:48 |
10500 |
10900 |
1.41 | 0.48 | 58 | 0.135 | 74 | 0.145 | 2.11 | 21.6 |
| mPEG1-b-PCL105a | — | 1:105 |
17000 |
18300 |
1.52 | 0.29 | 83 | 0.157 | 103 | 0.162 | 2.96 | 30.5 |
| mPEG2.7–(HPCL10)5.2b | 1:3:5 |
2:2.7:5.2 |
11300 |
12400 |
1.58 | 0.48 | 67 | 0.144 | 79 | 0.154 | 3.03 | 31.3 |
| mPEG2.6–(HPCL10)8.4b | 1:3:9 |
2:2.6:8.4 |
14800 |
15300 |
1.69 | 0.35 | 76 | 0.136 | 85 | 0.143 | 3.49 | 36.2 |
| mPEG2.6–(HPCL22)4.8b | 1:3:5 |
2:2.6:4.8 |
17700 |
18600 |
1.64 | 0.29 | 81 | 0.164 | 97 | 0.156 | 3.24 | 33.5 |
| mPEG5.3–(HPCL22)8.6b | 1:6:9 |
2:5.3:8.6 |
33000 |
35400 |
1.71 | 0.32 | 88 | 0.132 | 113 | 0.152 | 3.31 | 34.2 |
The molecular weight and polydispersity index (PDI) determined by GPC further confirm the successful synthesis of amphiphilic hyperbranched polymers. The results are listed in Table 1. The Mn data determined by GPC are consistent with those calculated on the basis of 1H NMR spectra. Normalized GPC curves of BPCL10, CPCL10, and mPEG2.7–(HPCL10)5.2 are shown in Fig. S1.† BPCL10 and CPCL10 are low molecular weights with unimodal molecular weight distributions. mPEG2.7–(HPCL10)5.2 is high molecular weight with unimodal molecular weight distribution. The result proves the successful synthesis of mPEG–HPCL.
Polymeric micelles of amphiphilic polymers were prepared by dialysis of a THF solution of the polymer against deionized water. Although amphiphilic hyperbranched polymers mPEG–HPCL contain the mixed hydrophobic (PCL) and hydrophilic (mPEG) segments, interior hyperbranched PCL and exterior linear mPEG can be considered as the separate segments on a molecular level. Therefore, amphiphilic hyperbranched polymers mPEG–HPCL will form core–shell polymeric micelles by self-assembly of many molecules in aqueous solution with HPCL as the core and mPEG as the shell. The micelle size and size distribution were determined by dynamic light scattering (DLS). The micelle sizes increase with the increasing of the molecular weight of hydrophobic HPCL core when the amount of hydrophilic mPEG is kept constant (Table 1). Furthermore, the increasing of the amount of hydrophilic mPEG also increases the micelle sizes. For comparison, amphiphilic linear block polymers mPEG1-b-PCL26 (where mPEG1 has a different molecular weight from mPEG in mPEG–HPCL, which is noted in Table 1) are used as a control in investigating micelle property, in vitro drug release, and in vitro cytotoxicity. The DLS histograms and the TEM images of the micelles prepared by mPEG1-b-PCL48 (where the number on the lower right corner of PCL stands for degree of polymerization of ε-caprolactone) and mPEG2.7–(HPCL10)5.2 are shown in Fig. 2. The measured values of DLS are 58 and 67 nm for mPEG1-b-PCL48 micelles and mPEG2.7–(HPCL10)5.2 micelles, respectively. Nanosized spherical particles are observed in the TEM images. The mean diameters of mPEG1-b-PCL48 micelles and mPEG2.7–(HPCL10)5.2 micelles based on the TEM images are about 40 and 50 nm, respectively.
 | ||
| Fig. 2 Size distribution profile determined by DLS for mPEG1-b-PCL48 micelles (A), mPEG2.7–(HPCL10)5.2 micelles (B), and TEM images of mPEG1-b-PCL48 micelles (C), mPEG2.7–(HPCL10)5.2 micelles (D). The micelles were produced at a concentration of approximately 0.5 mg mL−1 for DLS measurement. The micelles were negatively stained by phosphotungstic acid for TEM observation. | ||
Methotrexate was used as a model drug to test the properties of polymeric micelles. The drug-loaded micelles were prepared from the solutions of 10:1 (by weight) polymer/drug mixtures by a procedure similar to that for drug-free micelles. To determine the drug loading content (DLC) and entrapment efficiency (EE), the drug-loaded micelle solution was dried by rota-evaporation and the residue was dissolved in DMF to determine the drug concentration through measuring the UV absorbance at 303 nm. The results are summarized in Table 1. The drug-loaded micelles possess a bigger diameter than the corresponding drug-free micelles. When the amount of hydrophilic mPEG is similar, the drug loading capacity is dependent to both the amount and the length of hydrophobic PCL. Increasing the hydrophobic PCL amount or length is beneficial for improving the drug loading capacity. However, the longer hydrophobic PCL is disadvantageous to load the drug for the same molecular weight of hydrophobic HPCL core. The longer hydrophobic PCL has the fewer branching points and the decreasing of the branching point causes hyperbranched polymers to tend to block polymers. Hyperbranched polymers mPEG–HPCL possess better drug loading capacity than block polymers mPEG1-b-PCL. For example, mPEG2.7–(HPCL10)5.2 has an EE of 31.3%, which is approximately 45% higher than the 21.6% of mPEG1-b-PCL48. The hyperbranched structure of mPEG–HPCL increases the disorder and space between PCL chains, thus enhance the interaction of polymer chains with drug molecules.43
The drug release behavior was investigated in PBS (pH 7.4) by monitoring the drug amounts released from the drug-loaded micelle solution that was placed in a dialysis bag. The in vitro drug release profiles of methotrexate from mPEG1-b-PCL48 micelles and mPEG2.7–(HPCL10)5.2 micelles are shown in Fig. S2.† The drug release from mPEG1-b-PCL48 micelles is fast with a burst release. The drug release from mPEG2.7–(HPCL10)5.2 micelles is sustained for about a week. Approximately 78% and 51% of drug enwrapped in the micelles is release from mPEG1-b-PCL48 micelles and mPEG2.7–(HPCL10)5.2 micelles after 2 days, respectively. These results indicate hyperbranched polymers mPEG–HPCL are a superior biomaterial for drug controlled release.
The in vitro cytotoxicity of block polymers mPEG1-b-PCL and hyperbranched polymers mPEG–HPCL was investigated by MTT assay. The cell viability data at different concentrations of mPEG1-b-PCL48 and mPEG2.7–(HPCL10)5.2 are shown in Fig. S3.† In a wide range of polymer concentrations, the cell viability is higher than 85%, indicating block polymers mPEG1-b-PCL and hyperbranched polymers mPEG–HPCL have good biocompatibility.
Conclusions
In summary, we describe a facile strategy for synthesizing amphiphilic hyperbranched polymers mPEG–HPCL with biodegradable hyperbranched poly(ε-caprolactone) core from homologous AB2 macromonomer. mPEG–HPCL self-assemble to form polymeric micelles. The diameters of polymeric micelles determined by DLS are tens of nanometers and nanosized spherical particles are observed from the TEM images. Hyperbranched polymers mPEG–HPCL have better drug-loading property and more sustained drug release than the corresponding linear block polymers mPEG1-b-PCL. Therefore, mPEG–HPCL will be a very promising biodegradable and biocompatible biomaterial for drug controlled release.Acknowledgements
This work was financially supported by the National Basic Research Program of China (2011CB606202) and the National Natural Science Foundation of China (51403241).Notes and references
- N. Wiradharma, Y. Zhang, S. Venkataraman, J. L. Hedrick and Y. Y. Yang, Nano Today, 2009, 4, 302–317 CrossRef CAS.
- M. Licciardi, C. Scialabba, C. Sardo, G. Cavallaro and G. Giammona, J. Mater. Chem. B, 2014, 2, 4262–4271 RSC.
- A. Palanisamy and Q. P. Guo, RSC Adv., 2014, 4, 54752–54759 RSC.
- N. Engelhardt, A. Ernst, A. L. Kampmann and R. Weberskirch, Macromol. Chem. Phys., 2013, 214, 2783–2791 CrossRef CAS.
- B. E. McKenzie, F. Nudelman, P. H. H. Bomans, S. J. Holder and N. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256–10259 CrossRef CAS PubMed.
- I. Choi, S. T. Malak, W. N. Xu, W. T. Heller, C. Tsitsilianis and V. V. Tsukruk, Macromolecules, 2013, 46, 1425–1436 CrossRef CAS.
- S. Venkataraman, Z. A. Chowdhury, A. L. Lee, Y. W. Tong, I. Akiba and Y. Y. Yang, Macromol. Rapid Commun., 2013, 34, 652–658 CrossRef CAS PubMed.
- J. R. Finnegan, D. J. Lunn, O. E. C. Gould, Z. M. Hudson, G. R. Whittell, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2014, 136, 13835–13844 CrossRef CAS PubMed.
- G. Rizis, T. G. M. van de Ven and A. Eisenberg, Angew. Chem., Int. Ed., 2014, 53, 9000–9003 CrossRef CAS PubMed.
- H. C. Tsai, C. H. Chang, Y. C. Chiu, S. Y. Lin, C. P. Lin and G. H. Hsiue, Macromol. Rapid Commun., 2011, 32, 1442–1446 CrossRef CAS PubMed.
- G. Q. Cai, H. W. Zhang, P. Liu, L. Q. Wang and H. L. Jiang, Acta Biomater., 2011, 7, 3729–3737 CrossRef CAS PubMed.
- N. Ghasdian, D. M. A. Buzza, P. D. I. Fletcher and T. K. Georgiou, Macromol. Rapid Commun., 2015, 36, 528–532 CrossRef CAS PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- Z. L. Tyrrell, Y. Q. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128–1143 CrossRef CAS.
- H. Q. Hu, M. Gopinadhan and C. O. Osuji, Soft Matter, 2014, 10, 3867–3889 RSC.
- T. Rudolph, M. D. Luhe, M. Hartlieb, S. Norsic, U. S. Schubert, C. Boisson, F. D'Agosto and F. H. Schacher, ACS Nano, 2015, 9, 10085–10098 CrossRef CAS PubMed.
- K. H. Kim, J. C. Lee and J. Lee, Macromol. Biosci., 2008, 8, 339–346 CrossRef CAS PubMed.
- W. L. Zhang, Y. L. Li, L. X. Liu, Q. Q. Sun, X. T. Shuai, W. Zhu and Y. M. Chen, Biomacromolecules, 2010, 11, 1331–1338 CrossRef CAS PubMed.
- K. Yoon, H. C. Kang, L. Li, H. Cho, M. K. Park, E. Lee, Y. H. Bae and K. M. Huh, Polym. Chem., 2015, 6, 531–542 RSC.
- X. S. Fan, Z. G. Hu and G. W. Wang, RSC Adv., 2015, 5, 100816–100823 RSC.
- W. W. Zhang, D. L. Zhang, X. S. Fan, G. Y. Bai, Y. M. Guo and Z. G. Hu, RSC Adv., 2016, 6, 20761–20771 RSC.
- P. Tirino, C. Conte, M. Ordegno, R. Palumbo, F. Ungaro, F. Quaglia and G. Maglio, Macromol. Chem. Phys., 2014, 215, 1218–1229 CrossRef CAS.
- X. J. Wan, T. Liu and S. Y. Liu, Biomacromolecules, 2011, 12, 1146–1154 CrossRef CAS PubMed.
- L. T. Yan and X. M. Xie, Prog. Polym. Sci., 2013, 38, 369–405 CrossRef CAS.
- Y. Wang and S. M. Grayson, Adv. Drug Delivery Rev., 2012, 64, 852–865 CrossRef CAS PubMed.
- X. J. Zhang, J. A. Cheng, Q. R. Wang, Z. L. Zhong and R. X. Zhuo, Macromolecules, 2010, 43, 6671–6677 CrossRef CAS.
- X. J. Zhang, F. J. Chen, Z. L. Zhong and R. X. Zhuo, Macromol. Rapid Commun., 2010, 31, 2155–2159 CrossRef CAS PubMed.
- X. J. Zhang, Z. G. Zhang, Z. L. Zhong and R. X. Zhuo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2687–2696 CrossRef CAS.
- X. J. Zhang, Z. G. Zhang, X. Su, H. Dong, Z. L. Zhong and R. X. Zhuo, Macromol. Chem. Phys., 2015, 216, 1712–1717 CrossRef CAS.
- M. Hu, M. S. Chen, G. L. Li, Y. Pang, D. L. Wang, J. L. Wu, F. Qiu, X. Y. Zhu and J. Sun, Biomacromolecules, 2012, 13, 3552–3561 CrossRef CAS PubMed.
- D. Ernenwein, A. M. Vartanian and S. C. Zimmerman, Macromol. Chem. Phys., 2015, 216, 1729–1736 CrossRef CAS.
- Y. Huang, D. L. Wang, X. Y. Zhu, D. Y. Yan and R. J. Chen, Polym. Chem., 2015, 6, 2794–2812 RSC.
- M. J. Liu, N. Vladimirov and J. M. J. Frechet, Macromolecules, 1999, 32, 6881–6884 CrossRef CAS.
- X. H. Yu, J. Feng and R. X. Zhuo, Macromolecules, 2005, 38, 6244–6247 CrossRef CAS.
- P. G. Parzuchowski, M. Grabowska, M. Tryznowski and G. Rokicki, Macromolecules, 2006, 39, 7181–7186 CrossRef CAS.
- P. G. Parzuchowski, M. Jaroch, M. Tryznowski and G. Rokicki, Macromolecules, 2008, 41, 3859–3865 CrossRef CAS.
- M. Tryznowski, K. Tomczyk, Z. Fras, J. Gregorowicz, G. Rokicki, E. Wawrzynska and P. G. Parzuchowski, Macromolecules, 2012, 45, 6819–6829 CrossRef CAS.
- M. Trollsas and J. L. Hedrick, Macromolecules, 1998, 31, 4390–4395 CrossRef.
- M. Trollsas, B. Atthoff, H. Claesson and J. L. Hedrick, Macromolecules, 1998, 31, 3439–3445 CrossRef.
- J. Choi and S. Y. Kwak, Macromolecules, 2003, 36, 8630–8637 CrossRef CAS.
- C. Gottschalk and H. Frey, Macromolecules, 2006, 39, 1719–1723 CrossRef CAS.
- R. H. Lin, J. Castells and H. Rapoport, J. Org. Chem., 1998, 63, 4069–4078 CrossRef CAS.
- A. Kowalczuk, R. Trzcinska, B. Trzebicka, A. H. E. Muller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2014, 39, 43–86 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details. See DOI: 10.1039/c6ra08531h |
| This journal is © The Royal Society of Chemistry 2016 |