
Thermoresponsive dynamic covalent dendronized polymers†
Xiacong Zhang,
Jiatao Yan,
Wen Li* and
Afang Zhang*
Laboratory of Polymer Chemistry, Department of Polymer Materials, College of Materials Science and Engineering, Shanghai University, Materials Building Room 447, Nanchen Street 333, Shanghai 200444, China. E-mail: wli@shu.edu.cn; azhang@shu.edu.cn
First published on 28th April 2016
Abstract
Thermoresponsive dendronized polymers constituted with a polyacylhydrazone backbone pendanted with oligoethylene glycol (OEG)-based dendrons were synthesized through polycondensation of dialdehydes and diacylhydrazines. First (G1) and second (G2) generation OEG dendrons were selected for comparison to examine the dendritic effects. Inherited from OEG dendrons, these polymers showed characteristic thermoresponsive behavior. By virtue of the dynamic property of the acylhydrazone linkage, formation of these polymers are concentration and pH-dependent. A dimer from monoaldehyde and monoacylhydrazine was prepared as a model to support the remarkable macromolecular effects for enhanced structural stability of polyacylhydrazones. Constitutional stability of these polyacylhydrazones were also testified by switching solutions to strong acidic or basic conditions. The structural vibration was simply testified by addition of a new monomer as a competitor to dendronized polyacylhydrazones, thus, resulting in polymers with tunable phase transition temperatures. Furthermore, G1 dendrons were found to provide efficient shielding for acylhydrazones from an affinity to metal ions, but this shielding effect diminished and exhibited specific interactions with Cu2+ when the dendron collapsed.
Introduction
Dendronized polymers possessing linear backbones and tree-like pendants are one kind of unique hyperfunctional polymers with nanometer sizes and cylindrical shape.1 Their properties can be mediated through chemical structure modification at the polymer backbone, dendritic side chains and peripheral groups. Based on these attributes, these kinds of polymers have drawn significant interest and been utilized in many fields including drug delivery,2 catalytic supports,3 energy storage.4 Though dendronized polymers show great potential as candidates for functional materials, their syntheses are still challenging. There are three main approaches for synthesis of this novel class polymers: macromonomer route, “graft to” route and “graft from” route.1 Traditionally, these polymers were mostly constructed via covalent linkage which possess fixed structures and are lack of functionality. In recent years, supramolecular interactions including metal coordination, hydrogen bonding and host–guest interactions have been proved to be efficient and simple methodologies for construction of dendronized polymers.5 The formed polymers not only keep the structure characteristics from dendronized polymers but also show dynamic properties, which endow the polymers further responsive properties and enhanced functionalities. However, due to the relative weak bond energy of supramolecular interactions, these polymers are not stable in certain conditions.Recently, dynamic covalent chemistry (DCC) based on reversible covalent bonds has been used widely for preparing polymers with adaptive and self-healing properties.6 Dynamic covalent bond is a kind of stable reversible linkage which possesses capacity to be formed and broken under equilibrium control, affording the polymers not only with enhanced stability, but also with constitutional reversible features. Common dynamic covalent linkages include imines, Diels–Alder adducts, boronic esters and disulfide linkages.7 Till now, polymers with different architectures such as homopolymers, block copolymers, comb-like polymers, star-like polymers and cyclic polymers have been prepared via DCC. Specifically, dendrimers and hyperbranched polymers with stable and stimulus-triggered transformable structures based on DCC have also been developed. For example, a pH-triggered backbone cleavable hyperbranched polyacylhydrazone was prepared, which could be used for drug delivery.8 Similarly, hyperbranched polyoximes were also prepared which could self-assemble into nanoparticles with dual responsiveness to pH and temperature.9 Segmented hyperbranched copolymers with Diels–Alder linkage were reported, which could cleave into linear polymers with heating.10 Furthermore, dendrimers based on thermally reversible Diels–Alder reactions have been reported11 and for periphery modification.12 Recently, we prepared one class of dendronized polypeptides pendanted with OEG-based dendrons through either Schiff base bonds or oximes, which show tunable secondary structures and switchable thermoresponsiveness due to the DCC linkages.13 However, dendronized polymers constituted through DCC in their backbone are rarely reported.
In our previous work, a series of OEG-based first (G1) and second generation (G2) dendronized polymethacrylates were prepared via both covalent and supramolecular chemistry, which show fast and sharp phase transitions with negligible hysteresis in aqueous solutions.14 Their phase transition temperatures can be tuned in the range of 32–65 °C through changing dendron generation, interior, or peripheral units. Compared to covalent polymers, the dynamic features of supramolecular interactions through host–guest chemistry facilitate tunable phase transition temperatures by simply varying ratios of the guests of different hydrophilicities.15 However, thermally-induced aggregation causes decomposition of the host–guest complex which could affect their performances in potential applications. Therefore, we here report on OEG-based thermoresponsive dendronized polymers through dynamic covalent linkages to possess both high stability and dynamic features. Polyacylhydrazones pendanted with G1 or G2 OEG dendrons were synthesized through dynamic covalent reactions between dialdehydes and diacylhydrazines in the presence of acid catalyst. The effect of polymerization conditions on molecular weight was surveyed, and their thermoresponsive properties were investigated by UV/Vis spectroscopy. The dynamic constitutional feature of these dendronized polymers were examined by switching solution pH conditions and also through addition of competitive monomers with different hydrophilicity in order to mediate polymer thermoresponsiveness. Moreover, possible structure effect on binding of copper(II) ions by acylhydrazones was also investigated (Fig. 1).
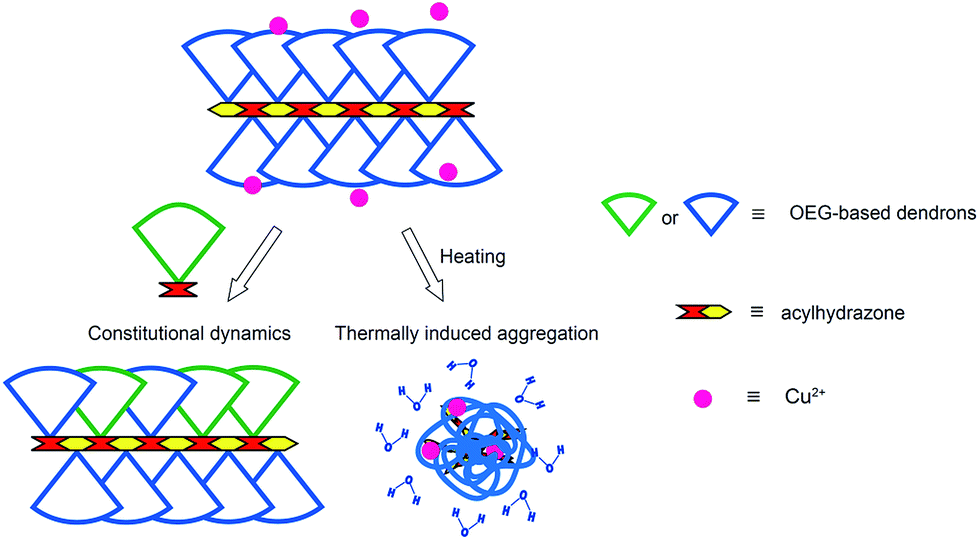 | ||
| Fig. 1 Illustration of thermoresponsive dendronized polymers constructed via dynamic covalent bonds showing constitutional dynamics and thermally controlled interactions with Cu2+. | ||
Results and discussion
Synthesis and characterization
To achieve dendronized polymers with dynamic acylhydrazone linkage in the backbone, OEG-based G1 and G2 monomers cored with dialdehydes or diacylhydrazines were firstly prepared. G1 monomers terminated with either methoxyl or ethoxyl groups were prepared to achieve G1 polymers with different inherited hydrophilicity. To achieve G2 polymers with a phase transition around physiological temperature, G2 monomers only with hydrophobic ethoxyl terminals were prepared. The detailed synthetic procedures for all monomers are delineated in Scheme 1. Starting from G1 and G2 dendron benzyl chlorides 1a, 1b and 3, Williamson etherification with dimethyl 5-hydroxyisophthalate afforded the corresponding di(ester)s 2a, 2c and 4a, respectively. These di(ester)s were reduced with LiAlH4 to benzyl di(alcohol)s 2b, 2d and 4b, respectively, which were then transferred through oxidation with pyridinium chlorochromate to corresponding dialdehydes G1MeA, G1EtA and G2EtA, respectively. The corresponding diacylhydrazines G1MeH, G1EtH and G2EtH were additionally prepared by hydrazinolysis of the respective di(ester)s 2a, 2c and 4a with hydrazine hydrate. Finally, G1 and G2 dendritic monomers carrying either aldehyde groups or acylhydrazine groups were effectively synthesized with an overall yield of 70% and 58%, respectively. G1MeA, G1EtA, G2EtH and G2EtA are all liquid, while G1MeH and G1EtH carrying acylhydrazine group are solid at room temperature. All new compounds were characterized by 1H NMR spectroscopy and high resolution mass spectrometry.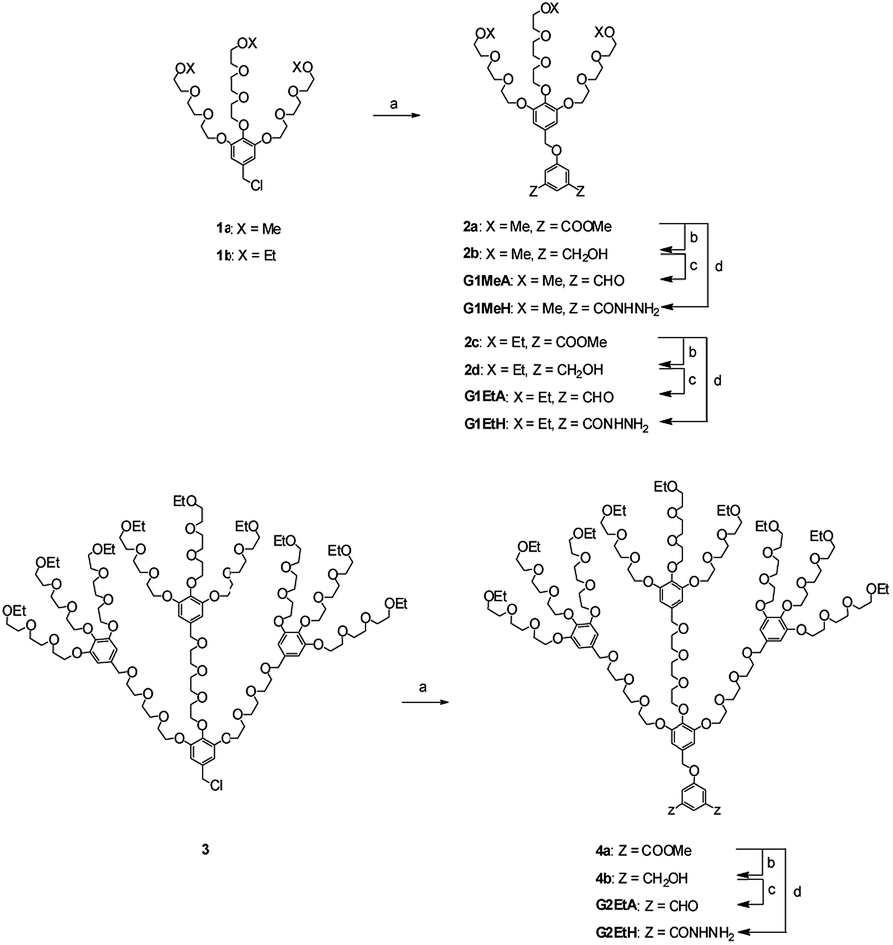 | ||
| Scheme 1 Synthetic procedures for the monomers. Reagents and conditions: (a) dimethyl 5-hydroxyisophthalate, KI, K2CO3, DMF, 80 °C (2a: 60%, 2c: 72%, 4a: 71%); (b) LiAlH4, THF, −5 °C to 25 °C (2b: 80%, 2d: 80%, 4b: 55%); (c) PCC, DCM, 0 °C to 25 °C (G1MeA: 63%, G1EtA: 70%, G2EtA: 84%); (d) hydrazine hydrate, MeOH (G1MeH: >99%, G1EtH: >99%, G2EtH: 84%). LiAlH4 = lithium aluminum hydride, PCC = pyridinium chlorochromate. | ||
Hydrazone formation is kinetically inertness under neutral conditions, but can be accelerated under acidic conditions (pH 2–5) or by using excess aniline as a nucleophilic catalyst.16 Herein, the effect of pH value on reactions between dendritic diacylhydrazine G1EtH and dialdehyde G1EtA (both carrying ethoxyl terminals) in D2O (3.1 mM) was selected to be followed by 1H NMR spectroscopy (Fig. 2). In neutral D2O solution, G1EtH and G1EtA only reacted at low degree (48%) as evidenced by presence of the well resolved signals from unreacted aldehyde (a–c) and acylhydrazine monomers (f and g). When pH value of the mixture decreased from 7.3 to 6.0 by addition of DCl, the proton signals turned to be broad except two sharp signals with decreased intensities from aldehyde and acylhydrazine moieties. The broadening of proton signals result from the characteristic short spin–spin relaxation times due to slow diffusion and reorientation, demonstrating the formation of polymer PEtG1. At pH 3.6 or below, the signals from aldehyde and acylhydrazine moieties at the aromatic region disappeared, suggesting high conversion of reactions. Notably, the proton signals from the polyacylhydrazone corresponding to both OEG segments (δ = 3.1–4.2 ppm and δ = 0.8–1.2 ppm) and aromatic moieties (δ = 6.0–8.3 ppm) are very broad. This is quite different from the OEG-based dendritic polymethacrylates reported in our previous work,14 where well-resolved signals due to the flexibility of polymer backbone were observed. This indicates the OEG-based dendronized polyacylhydrazones are quite rigid, mostly due to the semi-conjugated backbones. These broad proton signals remain even after adjusting solution pH above 7 with NaOD, proving the inertness of acylhydrazones to basic conditions in these OEG-based dendronized polymers. Moreover, obvious hydrolysis was not observed even at strong acidic conditions (pH 1.5 or below), indicating high stability of these dynamic polymers in aqueous solutions. For comparison, dimeric acylhydrazone 7 was prepared as a model from the corresponding acylhydrazine 5 and aldehyde 6 (for synthetic routes, see Scheme S1†), to examine stability of dendronized acylhydrazones at acidic conditions by 1H NMR spectroscopy (Fig. 3). The yield of dendronized acylhydrazone was calculated by comparing the proton intensities of aldehyde moiety at 9.7 ppm from compound 6 and that of acylhydrazone moiety at 8.2 ppm from compound 7. Similar to polymeric system, yield of dimeric acylhydrazone is quite high (98%) at weak acid condition (pH 4.7), however, the aldehyde signals at the aromatic region reappear and the yield of acylhydrazone decrease to 56% at pH 0.85. These results indicate polyacylhydrazones carrying OEG-based dendrons in the pendants show a higher tolerance toward acid than the dimeric counterpart. We ascribe the significant difference in stability between polymeric and monomeric acylhydrazones to the macromolecular effect. On dendronized polyacylhydrazones, cylindrical geometry of the polymers help the densely covered OEG dendrons in the pendants to shield the backbone from protonation even at strong acidic conditions. This phenomenon is similar to our previous finding in OEG-based dendronized polymethacrylates, where protonation of solvatochromic dyes were prohibited due to the shielding effect from the surrounded OEG dendrons.17
 | ||
| Fig. 2 1H NMR spectra from mixtures of G1EtH and G1EtA in D2O with different pH values. [G1EtH] = [G1EtA] = 3.1 mM. | ||
 | ||
| Fig. 3 1H NMR spectra from mixtures of 5 and 6 in D2O at different pH conditions. C = 5 mM. | ||
Based on above results, the polymerization of all monomers were carried out in acidic buffer solutions to efficiently afford polyacylhydrazones. To examine effect of monomer concentration and pH value on polymer molecular weight, the polymerization of G1MeH and G1MeA were also conducted with different monomer concentrations and in different pH solutions. All monomers are soluble in water at room temperature. The dynamic polycondensation of equivalent quantities of diacylhydrazine and dialdehyde monomers were carried out at room temperature and all polymerization proceeded homogeneously over 48 h. The resulting solutions were lyophilized and then purified by silica gel column chromatography to afford the products. The detailed polymerization conditions and typical results are summarized in Table 1. All polymer weight-average molecular weights (Mws) were determined by GPC-LS with DMF as the eluent and are in the range of 33–215 × 103 with polydispersity (PDI) less than 1.7. With increasing initial concentration of monomers G1MeH and G1MeA from 2 to 130 mM, Mws of the resulted polymers PMeG1 increased significantly from 33 × 103 to 125 × 103. While changing solution pH value from 3.64 to 2.00, Mws increased from 54 × 103 to 59 × 103, suggesting molecular weights of the polymers are less relevant at acidic condition to solution pH values. Moreover, copolymers PEtHMeAG1 and PEtAMeHG1 carrying methoxyl and ethoxyl terminals alternatively were obtained by polymerization of G1EtH with G1MeA and G1EtA with G1MeH, respectively. Surprisingly, PEtHMeAG1 was obtained with the highest molecular weight, which may be due to the complementarity in hydrophilicity of the comonomers. All polymers obtained are solids except second generation (G2) dendronized polyacylhydrazones (PEtG2) which is highly viscous. These polymers are well soluble in water at low temperature as well as in common organic solvent, such as CHCl3, methanol and DMF. The morphologies of polymers PEtG1 and PEtG2 were also investigated by atomic force microscopy (AFM) after spin-coated on mica (Fig. 4). It is obvious that the single molecular chains from G2 polymers are thicker and show higher height when compared to the G1 polymer chains. To the best of our knowledge, this is the first example to directly visualize the individual dynamic covalent polymer chains on substrates.
| Samples | pH | [M] (mM) | Yield (%) | GPC-LSa | Tcb (°C) | |
|---|---|---|---|---|---|---|
| Mw (×10−3) | PDI | |||||
| a Determined by GPC with light-scattering detector (DMF as eluent containing 0.1 wt% LiBr).b Cloud point temperatures (Tcs) of the polymers were determined as the temperature at 50% of the initial transmittance at λ = 700 nm. | ||||||
| PEtG1 | 3.64 | 20 | 78 | 45 | 1.24 | 37.7 |
| PMeG1 (a) | 3.64 | 20 | 77 | 54 | 1.15 | 75.6 |
| PMeG1 (b) | 3.64 | 2 | 76 | 33 | 1.16 | — |
| PMeG1 (c) | 3.64 | 130 | 50 | 125 | 1.27 | — |
| PMeG1 (d) | 2.00 | 20 | 74 | 59 | 1.29 | — |
| PEtG2 | 3.64 | 20 | 78 | 97 | 1.13 | 40.5 |
| PEtHMeAG1 | 3.64 | 20 | 43 | 215 | 1.70 | 49.5 |
| PEtAMeHG1 | 3.64 | 20 | 64 | 41 | 1.18 | 51.4 |
 | ||
| Fig. 4 AFM height images of PEtG1 and PEtG2 on mica. Samples were prepared from chloroform solutions (2 mg L−1) by spin-coating with a speed of 3000 rpm. | ||
Thermoresponsive properties and constitutional dynamics
These polymers are well water-soluble at room temperature, but collapse from water and their aqueous solutions turn into turbid when heated above their cloud points (Tcs). The thermally-induced collapse processes are fully reversible as the polymer solutions become clear and homogeneous again after cooled down to room temperature. Therefore, thermoresponsive behavior of these polymers were investigated and Tcs were determined by turbidity measurements using UV/Vis spectroscopy. Typical phase transition curves are shown in Fig. 5. The Tcs for PEtG1, PMeG1, and PEtG2 are 37.3, 75.6 and 41.2 °C, respectively. The hysteresis span less than 3 °C for G1 polymers and 1 °C for G2 polymer. It deserves to point out that dendritic polymers PEtG1 and PEtG2 show Tcs around physiological temperature, which may pave a way for bioapplications. This is in contrast to the previous report that polyacylhydrazones carrying linear OEG pendants display thermoresponsive behavior with phase transition temperatures above 75 °C.18 Furthermore, when compared with OEG-based covalent dendronized polymethacrylates, the phase transition temperatures of these polyacylhydrazones show obvious increase of about 4 °C.14a This should be contributed from the hydrophilicity of polyacylhydrazone backbone. Tcs for copolymers PEtHMeAG1 (from hydrophobic G1EtH and hydrophilic G1MeA) and PEtAMeHG1 (from hydrophilic G1MeH and hydrophobic G1EtA) were found to be 49.5 °C and 51.4 °C, respectively, which are in between the Tcs of PEtG1 and PMeG1. This indicates that dynamic covalent chemistry can be an efficient tool to tune the polymer hydrophilicity as well as their phase transition temperatures through copolymerization of two dendritic monomers of different hydrophilicity. | ||
| Fig. 5 Plots of transmittance vs. temperature for 0.25 wt% aqueous solutions of PEtG1, PEtG2, PMeG1 and PEtHMeAG1. Heating rate = 0.2 °C min−1. | ||
A characteristic feature for acylhydrazone is its ability to undergo exchange reaction under acidic conditions. The dynamic character of the polymers was thus investigated through the component-exchange experiments by further addition of diacylhydrazine monomers with different hydrophilicities to the formed polyacylhydrazones. After component-exchange reactions between the added monomers and the polymers, the Tcs of the polymers are expected to be changed due to the change of polymers hydrophilicity.19 Before addition of monomers, the Tcs of PEtG1, PEtG2 and PMeG1 in buffer (0.25 wt%) at pH 1.6 were determined to be 37.0, 41.1 and 73.3 °C, respectively (for their phase transition curves, see Fig. S1†). After addition of more hydrophobic ethoxyl terminated monomers G1EtH to the buffer of PMeG1, the Tc decreases significantly from 73.3 °C to 56.1 °C in 5 days. The component-exchange process was followed by 1H NMR spectroscopy with time. As shown in Fig. 6a, proton signals from methoxyl terminated groups of monomer G1MeH increased obviously after 5 days, which indicate that the exchange reaction occurred and the free G1MeH monomers were released. While after addition of more hydrophilic methoxyl terminated monomer G1MeH into the buffer of more hydrophobic ethoxyl terminated polymer PEtG1 and PEtG2, respectively, both of their Tcs change less than 2 °C as shown in Fig. 6b. These results suggest more hydrophobic monomers show stronger competitiveness which can be efficiently incorporated into the polymer chains of PMeG1 constructed with more hydrophilic monomers, resulting in a decrease of the over-all hydrophilicity of the polymers. However, more hydrophilic monomers show weaker competitiveness, which is hardly to be incorporated into PEtG1 or PEtG2 constructed with more hydrophobic monomers.
 | ||
| Fig. 6 (a) 1H NMR spectra of PMeG1 and G1EtH aqueous solutions (pH = 1.6) at different time intervals (1 d, 5 d, 7 d); (b) evolution of the Tcs of PMeG1, PEtG1 and PEtG2 after addition of an equivalent of new monomers. Heating rate = 0.2 °C min−1. | ||
Switchable binding to copper ions
Acylhydrazone are able to act as ligands for Cu2+ ions.20 As the third most abundant transition metal ions, Cu2+ and its complexes play vital roles in both industrial applications and biological systems. Therefore, the selective detection of Cu2+ is essential in both environmental science and biological systems. The interactions between these dendronized polyacylhydrazones and Cu2+ was thus investigated with temperature-varied UV/Vis spectroscopy in aqueous solutions. The absorption spectrum of PEtG1 solution at room temperature without the metal ion exhibit two intense absorption bands at 208 and 300 nm (Fig. S2†). The peak from 250 to 400 nm is attributed to acylhydrazone moieties. When the solution temperature increased to 45 °C which is above the Tc for PEtG1, the absorbance at 300 nm increase slightly. When an equivalent of Cu2+ to acylhydrazone moieties was added to the polymer solution, the absorption spectra remain unchanged at room temperature, indicating the interaction between acylhydrazone moieties with Cu2+ is prohibited somehow. However, when solution temperature increases above its Tc, absorption at 302 nm significantly decreased, and a new absorption band appeared at 370 nm with a clear isosbestic point at 333 nm (Fig. 7a), indicating that, at elevated temperature, OEG collapse facilitates the interaction between acylhydrazone moieties with Cu2+ within the metrics of PEtG1. However, this thermally-induced spectrum change is irreversible when cooling to room temperature, suggesting the strong interactions between acylhydrazones and Cu2+. Above results suggest that the shielding of OEG-based dendron pendants could efficiently weaken the interactions between acylhydrazones in the polymer backbone with Cu2+ below phase transition temperature, while the shielding ability decreases after dehydration. Therefore, the thermally induced phase transitions can effectively switch the interactions between acylhydrazone units in polymer with Cu2+. To investigate the selectivity of PEtG1 to metal ions, 1 equiv. of Na+, Ni2+, Li+, Zn2+, Ba2+, Ni2+ were also added to the polymer solution above the Tc (90 °C), respectively. From UV spectra in Fig. 7b, these cations do not induce discernible changes in absorption, therefore, PEtG1 exhibits an excellent selectivity toward Cu2+ binding. In contrast to PEtG1, PEtG2 only show minor change even above its Tcs with an isosbestic point at 356 nm when addition of 1 equiv. of Cu2+ (Fig. S3†), suggesting shielding of interaction between acylhydrazones and Cu2+ from G2 dendrons is too strong to be broken by thermally-induced collapse.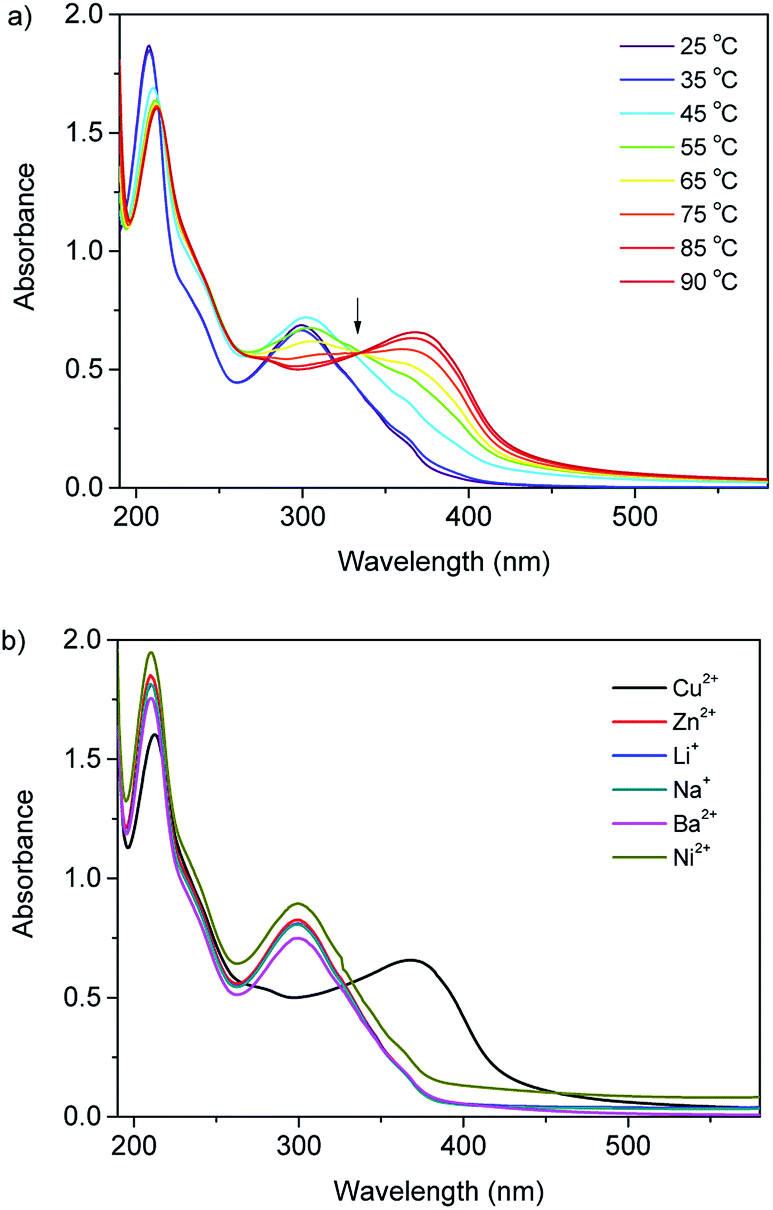 | ||
Fig. 7 (a) UV/Vis spectra of PEtG1 aqueous solutions and CuSO4 at different temperatures, [–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–] N–]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Cu2+] = 1:1, [PEtG1] = 39 μg mL−1. (b) UV/Vis spectra of PEtG1 aqueous solutions in the presence of 1 equivalent of different metal ions at 90 °C. :[Cu2+] = 1:1, [PEtG1] = 39 μg mL−1. (b) UV/Vis spectra of PEtG1 aqueous solutions in the presence of 1 equivalent of different metal ions at 90 °C. | ||
Conclusions
A series of OEG-based dynamic covalent dendronized polymers with polyacylhydrazone backbone were efficiently prepared. The weight-average molecular weights (Mws) of these polymers are influenced by the monomer concentrations and complementarity of monomer hydrophilicity. These dynamic polymers show remarkable macromolecular effect in enhancing high constitutional stability even at strong acidic solutions (pH 1.5 or below). All polymers show thermoresponsive properties, and their Tcs can be varied between 37.3 and 75.6 °C by changing peripheral groups of the dendrons. Taking advantages of the dynamic covalent linkages, the thermoresponsiveness of these polymers can also be mediated through addition of new dendritic monomers with different hydrophilicity (carrying either methoxyl or ethoxyl terminals), thus resulting in new polymers with different phase transition temperatures. Remarkably, G1 polymer shows switchable specific interaction with Cu2+ mediated with the phase transitions. These OEG-based dynamic covalent dendronized polymers with tunable phase transition temperatures and thermally controlled interactions with Cu2+ may provide a new platform for using as biomedical and controllable catalytic materials.Experimental
Materials
Dimethyl 5-hydroxyisophthalate, methyl 3-hydroxybenzoate, hydrazine hydrate, pyridinium chlorochromate (PCC), lithium aluminum hydride (LiAlH4) were purchased from TCI (Japan). Potassium carbonate (K2CO3) and KI were purchased from Sinopharm Chemical Reagent Co., Ltd. Dichloromethane (DCM) was dried over CaH2. Tetrahydrofuran (THF) was pre-dried over sodium hydride (NaH) and then refluxed over LiAlH4 before use. Potassium carbonate (K2CO3) was pre-dried at 120 °C in vacuo 24 h and KI was pre-dried at 80 °C in vacuo 24 h. Other reagents and solvents were purchased at reagent grade and used without further purification. Merck KGaA TLC silica gel 60 F254 were used for thin-layer chromatography (TLC) analysis. Silica gel 60 M (Macherey-Nagel, 0.04–0.063 mm, 200–300 mesh) was used as the stationary phase for column chromatography.Instrumentation and measurements
1H and 13C NMR spectra were recorded on a Bruker AV 500 (1H: 500 MHz, 13C: 125 MHz) spectrometer and chemical shifts are reported as δ values (ppm) relative to internal Me4Si. Gel permeation chromatography (GPC) measurements were carried out on a Waters GPC e2695 instrument with 3 column set (Styragel HR3 + HR4 + HR5) equipped with refractive index detector (Waters 2414), and DMF (containing 1 g L−1 LiBr) as eluent at 45 °C. Multi-angle light scattering detector (Wyatt Technology Corporation, Dawn EOS 243-E) was used for some of the measurements. The calibration was performed with poly(methyl methacrylate) standards in the range of Mp = 2580–981000 (Polymer Standards Service-USA Inc.). UV/Vis turbidity measurements were carried out on a PE UV/Vis spectrophotometer (Lambda 35) equipped with a thermo-controlled bath. Polymer aqueous solutions were placed in the spectrophotometer (path length 1 cm) and heated or cooled at a rate of 0.2 °C min−1. The absorptions of the solution at λ = 700 nm were recorded per 5 s. The turbid point (Tc) is determined the one at which the transmittance at λ = 700 nm had reached 50% of its initial value. The pH value was measured with a Mettler-Toledo Seven Compact 220 pH meter and an InLab Flex-Micro semi-micro electrode (after 4 points calibration at pH 10.00, 7.00, 4.01 and 2.00). Atomic force microscope (AFM) measurements were performed on a Bruker Multi-Mode 8 microscope with an “E” scanner (scanning range 10 μm × 10 μm) and operated in peak force mode at room temperature in air. Bruker silicon tip was attached on nitride lever cantilevers (T: 650 nm; L: 115 μm; W: 25 μm; f0: 70 kHz; k: 0.4 N m−1). Samples were prepared from chloroform solutions (2 mg L−1) by spin-coating with a speed of 3000 rpm. The scanning rate was at line frequency of 0.977 Hz. The images analyses were performed by using Nanoscope Analysis software.
Synthesis
Compound 2a. According to general procedure A from dimethyl 5-hydroxyisophthalate (0.82 g, 3.90 mmol), 1a (2.32 g, 3.87 mmol), KI (0.39 g, 2.50 mmol), K2CO3 (1.47 g, 11.35 mmol) and dry DMF, 2a was yielded as a yellow oil (1.79 g, 60%). 1H NMR (CDCl3): δ = 3.38–3.39 (d, 9H, CH3), 3.55–3.57 (m, 6H, CH2), 3.64–3.69 (m, 12H, CH2), 3.73–3.76 (m, 6H, CH2), 3.80–3.82 (t, 2H, CH2), 3.86–3.88 (t, 4H, CH2), 3.96 (s, 6H, CH3), 4.16–4.20 (m, 6H, CH2), 5.04 (s, 2H, CH2), 6.69 (s, 2H, CH), 7.83–7.84 (d, 2H, CH), 8.31–8.32 (t, 1H, CH). 13C NMR (CDCl3): δ = 52.37, 58.90, 68.78, 69.63, 70.40, 70.43, 70.48, 70.58, 70.71, 71.83, 71.68, 72.25, 107.05, 120.07, 123.18, 131.41, 131.75, 152.74, 158.58, 165.95. HRMS (MALDI): m/z calcd for C38H58O17 [M + Na]+: 809.3544. Found: 809.3566.
Compound 2b. According to general procedure B from LiAlH4 (0.21 g, 5.46 mmol), 2a (1.79 g, 2.27 mmol) and dry THF, 2b was yielded as a bright yellow oil (1.33 g, 80%). 1H NMR (CDCl3): δ = 3.38–3.39 (d, 9H, CH3), 3.54–3.57 (m, 6H, CH2), 3.64–3.67 (m, 12H, CH2), 3.72–3.74 (m, 6H, CH2), 3.79–3.81 (t, 2H, CH2), 3.84–3.8 (t, 4H, CH2), 4.14–4.18 (m, 6H, CH2), 4.65 (s, 4H, CH2), 5.00 (s, 2H, CH2), 6.68 (s, 2H, CH), 6.92 (s, 2H, CH), 6.93 (s, 1H, CH).
Compound G1MeA. According to general procedure C from PCC (1.02 g, 4.73 mmol), 2b (1.33 g, 1.82 mmol) and dry DCM, G1MeA was yielded as a bright yellow oil (0.83 g, 63%). 1H NMR (CDCl3): δ = 3.38–3.39 (d, 9H, CH3), 3.55–3.57 (m, 6H, CH2), 3.65–3.69 (m, 12H, CH2), 3.73–3.76 (m, 6H, CH2), 3.80–3.82 (t, 2H, CH2), 3.86–3.88 (t, 4H, CH2), 4.16–4.20 (m, 6H, CH2), 5.09 (s, 2H, CH2), 6.70 (s, 2H, CH), 7.74 (s, 2H, CH), 8.00 (s, 1H, CH), 10.07 (s, 2H, HC
O). 13C NMR (CDCl3): δ = 58.90, 68.85, 69.62, 70.42, 70.44, 70.46, 70.59, 70.62, 70.71, 71.83, 71.86, 72.26, 107.17, 120.12, 124.27, 130.92, 138.32, 138.37, 152.83, 159.70, 190.79. HRMS (MALDI): m/z calcd for C36H54O15 [M + Na]+: 749.3347. Found: 749.3355.
Compound G1MeH. According to general procedure D from 2a (0.62 g, 7.88 mmol), hydrazine hydrate (1.54 mL, 37.72 mmol) and anhydrous MeOH (6 mL), G1MeH was yielded as a bright yellow solid (0.62 g, >99%). 1H NMR (DMSO-d6): δ = 3.22–3.24 (d, 9H, CH3), 3.50–3.53 (m, 12H, CH2), 3.57–3.60 (m, 6H, CH2), 3.66–3.68 (t, 2H, CH2), 3.73–3.75 (t, 4H, CH2), 4.01–4.03 (t, 2H, CH2), 4.10–4.11 (t, 4H, CH2), 4.53 (s, 4H, NH2), 5.07 (s, 2H, CH2), 6.80 (s, 2H, CH), 7.56–7.57 (d, 2H, CH), 7.87 (s, 1H, CH), 9.81 (s, 2H, NH). 13C NMR (CDCl3): δ = 58.06, 58.86, 58.91, 58.96, 68.61, 68.68, 69.66, 70.07, 70.35, 70.38, 70.43, 70.49, 70.52, 70.53, 70.60, 70.62, 70.66, 70.73, 71.80, 71.83, 71.88, 72.21, 74.60, 106.60, 106.99, 117.06, 131.86, 137.59, 152.48, 152.52, 152.78. HRMS (MALDI): m/z calcd for C36H58N4O15 [M + Na]+: 809.3786. Found: 809.3791.
Compound 2c. According to general procedure A from dimethyl 5-hydroxyisophthalate (0.49 g, 2.35 mmol), 1b (1.40 g 2.14 mmol), KI (0.23 g, 1.41 mmol), K2CO3 (0.89 g, 6.41 mmol) and dry DMF, 2c was yielded as a colorless oil (1.28 g, 72%). 1H NMR (CDCl3): δ = 1.20–1.24 (m, 9H, CH3), 3.51–3.56 (m, 6H, CH2), 3.59–3.61 (m, 6H, CH2), 3.65–3.69 (m, 12H, CH2), 3.73–3.76 (m, 6H, CH2), 3.80–3.82 (t, 2H, CH2), 3.86–3.88 (t, 4H, CH2), 3.96 (s, 6H, CH3), 4.16–4.20 (m, 6H, CH2), 5.04 (s, 2H, CH2), 6.69 (s, 2H, CH), 7.83–7.84 (d, 2H, CH), 8.31 (s, 1H, CH). 13C NMR (CDCl3): δ = 15.08, 52.35, 66.51, 68.82, 69.63, 69.73, 69.76, 70.40, 70.43, 70.47, 70.58, 70.60, 70.73, 72.26, 107.07, 120.05, 123.16, 131.37, 131.74, 138.20, 152.76, 158.58, 165.91. HRMS (MALDI): m/z calcd for C41H64O17 [M + Na]+: 851.4030. Found: 851.4036.
Compound 2d. According to general procedure B from LiAlH4 (0.22 g, 5.79 mmol), 2c (2.00 g, 2.41 mmol) and dry THF, 2d was yielded as a colorless oil (1.33 g, 80%). 1H NMR (CDCl3): δ = 1.17–1.21 (m, 9H, CH3), 2.36 (s, 2H, OH), 3.48–3.53 (m, 6H, CH2), 3.55–3.58 (m, 6H, CH2), 3.61–3.65 (m, 12H, CH2), 3.69–3.71 (m, 6H, CH2), 3.76–3.78 (t, 2H, CH2), 3.81–3.83 (t, 4H, CH2), 4.12–4.16 (m, 6H, CH2), 4.62 (s, 4H, CH2), 4.96 (s, 2H, CH2), 6.65 (s, 2H, CH), 6.89 (s, 2H, CH), 6.90 (s, 1H, CH).
Compound G1EtA. According to general procedure C from PCC (0.28 g, 1.30 mmol), 2d (0.39 g, 5.04 mmol) and dry DCM, G1EtA was yielded as a colorless oil (0.27 g, 70%). 1H NMR (CDCl3): δ = 1.20–1.24 (m, 9H, CH3), 3.51–3.56 (m, 6H, CH2), 3.59–3.62 (m, 6H, CH2), 3.65–3.69 (m, 12H, CH2), 3.73–3.76 (m, 6H, CH2), 3.81–3.83 (t, 2H, CH2), 3.87–3.89 (t, 4H, CH2), 4.16–4.20 (m, 6H, CH2), 5.09 (s, 2H, CH2), 6.70 (s, 2H, CH), 7.74 (d, 2H, CH), 8.00 (s, 1H, CH), 10.08 (s, 2H, HC
O). 13C NMR (CDCl3): δ = 15.10, 66.55, 68.85, 69.63, 69.74, 70.45, 70.48, 70.58, 70.61, 70.75, 72.29, 107.18, 120.18, 124.30, 130.94, 138.34, 152.85, 159.73, 190.79. HRMS (MALDI): m/z calcd for C39H60O15 [M + Na]+: 791.3825. Found: 791.3825.
Compound G1EtH. According to general procedure D from 2c (0.2 g, 0.24 mmol), hydrazine hydrate (0.5 mL, 10.29 mmol) and anhydrous MeOH (4 mL), G1EtH was yielded as a white solid (0.2 g, >99%). 1H NMR (DMSO-d6): δ = 1.07–1.11 (m, 9H, CH3), 3.39–3.48 (m, 24H, CH2), 3.49–3.54 (m, 6H, CH2), 3.67–3.69 (t, 2H, CH2), 3.74–3.76 (t, 4H, CH2), 4.01–4.03 (t, 2H, CH2), 4.10–4.12 (t, 4H, CH2), 4.54 (s, 4H, NH2), 5.07 (s, 2H, CH2), 6.80 (s, 2H, CH), 7.57 (d, 2H, CH), 7.89 (s, 1H, CH), 9.80 (s, 2H, NH). 13C NMR (CDCl3): δ = 15.02, 15.06, 66.54, 68.60, 69.63, 69.66, 69.69, 70.09, 70.34, 70.48, 70.50, 70.53, 70.64, 72.21, 106.66, 116.94, 117.81, 131.82, 134.03, 137.62, 152.48, 158.73, 166.67. HRMS (MALDI): m/z calcd for C39H64N4O15 [M + Na]+: 851.4287. Found: 851.4260.
Compound 4a. According to general procedure A from dimethyl 5-hydroxyisophthalate (34.0 mg, 160.7 μmol), 3 (0.26 g, 107.1 μmol), KI (20.0 mg, 117.8 μmol), K2CO3 (99.0 mg, 717.7 μmol) and dry DMF, 4a was yielded as a colorless oil (0.20 g, 71%). 1H NMR (CDCl3): δ = 1.21–1.24 (m, 27H, CH3), 3.52–3.56 (m, 18H, CH2), 3.59–3.75 (m, 96H, CH2), 3.79–3.88 (m, 24H, CH2), 3.96 (s, 6H, CH3), 4.13–4.20 (m, 24H, CH2), 4.45 (s, 4H, CH2), 4.60 (s, 2H, CH2), 5.04 (s, 2H, CH2), 6.60 (s, 6H, CH), 6.70 (s, 2H, CH), 7.84 (d, 2H, CH), 8.32 (s, 2H, CH). 13C NMR (CDCl3): δ = 15.09, 52.38, 58.02, 66.53, 68.71, 68.78, 69.26, 69.29, 69.63, 69.66, 69.74, 69.75, 70.41, 70.46, 70.57, 70.59, 70.71, 72.22, 73.13, 107.04, 107.13, 120.07, 123.20, 131.44, 131.77, 137.63, 152.52, 152.74, 158.60, 165.94. HRMS (MALDI): m/z calcd for C128H214O53 [M + Na]+: 2622.3952. Found: 2622.3943.
Compound 4b. According to general procedure B from LiAlH4 (82.3 mg, 2.17 mmol), 4a (1.41 g, 542.1 μmol) and dry THF, 4b was yielded as a yellow oil (0.76 g, 55%).
Compound G2EtA. According to general procedure C from PCC (0.13 g, 601.2 μmol), 4b (0.51 g, 200.4 μmol) and dry DCM, G2EtA was yielded as a bright yellow oil (0.50 g, 84%). 1H NMR (CDCl3): δ = 1.20–1.24 (m, 27H, CH3), 3.51–3.88 (m, 138H, CH2), 4.12–4.20 (m, 24H, CH2), 4.45 (s, 6H, CH2), 5.08 (s, 2H, CH2), 6.58 (s, 6H, CH), 6.70 (s, 2H, CH), 7.73 (d, 2H, CH), 8.00 (s, 1H, CH), 10.07 (s, 2H, HC
O). 13C NMR (CDCl3): δ = 15.10, 52.71, 66.54, 68.84, 69.26, 69.29, 69.64, 69.66, 69.74, 69.76, 70.42, 70.46, 70.55, 70.57, 70.60, 70.63, 70.72, 72.23, 72.29, 73.14, 73.15, 107.04, 107.27, 120.16, 124.30, 131.02, 133.70, 133.72, 137.64, 138.34, 138.38, 152.53, 152.82, 159.71, 190.82. HRMS (MALDI): m/z calcd For C126H210O51 [M + Na]+: 2562.3781. Found: 2562.3731.
Compound G2EtH. According to general procedure D from 4a (0.75 g, 288.4 μmol), hydrazine hydrate (0.9 mL, 18.52 mmol) and anhydrous MeOH (2 mL), and purification by column chromatography afforded the product. G2EtA was yielded as a bright yellow oil (0.63 g, 84%). 1H NMR (DMSO-d6): δ = 1.06–1.11 (m, 27H, CH3), 3.38–3.73 (m, 138H, CH2), 3.97–4.11 (m, 24H, CH2), 4.37 (s, 6H, CH2), 4.52 (s, 4H, NH2), 5.06 (s, 2H, CH2), 6.61 (s, 6H, CH), 6.79 (s, 2H, CH), 7.57 (d, 2H, CH), 7.89 (s, 1H, CH), 9.79 (s, 2H, NH). 13C NMR (CDCl3): δ = 15.06, 15.08, 66.55, 68.58, 68.65, 68.88, 69.31, 69.32, 69.66, 69.70, 69.71, 69.73, 69.74, 70.16, 70.38, 70.40, 70.48, 70.52, 70.55, 70.58, 70.60, 70.69, 70.70, 72.19, 72.21, 73.09, 73.12, 76.92, 106.84, 106.94, 107.43, 117.05, 117.39, 131.57, 133.73, 133.75, 134.21, 137.42, 137.53, 138.21, 152.39, 152.47, 152.64, 158.82, 166.61. HRMS (MALDI): m/z calcd for C126H214N4O51 [M + Na]+: 2622.4208. Found: 2622.4167.
Acknowledgements
We sincerely thank Dr Hongmei Deng from the Instrumental Analysis and Research Center of Shanghai University for NMR measurements. This work was financially supported by the National Natural Science Foundation of China (No. 21374058, 21474060, and 21574078) and the Shanghai Rising-Star Program (No. 16QA1401800).Notes and references
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- X. Yang, H. Shang, C. Ding and J. Li, Polym. Chem., 2015, 6, 668–680 RSC.
- (a) Y.-Y. Huang, X. Yang, Y. Feng, F. Verpoort and Q.-H. Fan, J. Mol. Catal. A: Chem., 2014, 393, 150–155 CrossRef CAS; (b) G. Fuhrmann, A. Grotzky, R. Lukić, S. Matoori, P. Luciani, H. Yu, B. Zhang, P. Walde, A. D. Schlüter, M. A. Gauthier and J.-C. Leroux, Nat. Chem., 2013, 5, 582–589 CrossRef CAS PubMed.
- M. M. Nielsen, I. Dimitrov, S. Takamuku, P. Jannasch, K. Jankova and S. Hvilsted, Fuel Cells, 2013, 13, 342–354 CrossRef CAS.
- J. Yan, W. Li and A. Zhang, Chem. Commun., 2014, 50, 12221–12233 RSC.
- (a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (b) A. Herrmann, Chem. Soc. Rev., 2013, 43, 1899–1933 RSC; (c) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634 RSC; (d) R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- (a) Y. Zhang and M. Barboiu, Chem. Rev., 2016, 116, 809–834 CrossRef CAS PubMed; (b) J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS; (c) T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- L. Zhu, C. Tu, B. Zhu, Y. Su, Y. Pang, D. Yan, J. Wu and X. Zhu, Polym. Chem., 2011, 2, 1761–1768 RSC.
- Y. Jin, L. Song, D. Wang, F. Qiu, D. Yan, B. Zhu and X. Zhu, Soft Matter, 2012, 8, 10017–10025 RSC.
- H. Sun, C. P. Kabb and B. S. Sumerlin, Chem. Sci., 2014, 5, 4646–4655 RSC.
- (a) J. R. McElhanon and D. R. Wheeler, Org. Lett., 2001, 3, 2681–2683 CrossRef CAS PubMed; (b) M. L. Szalai, D. V. McGrath, D. R. Wheeler, T. Zifer and J. R. McElhanon, Macromolecules, 2007, 40, 818–823 CrossRef CAS.
- (a) C. Kim, H. Kim and K. Park, J. Organomet. Chem., 2003, 667, 96–102 CrossRef CAS; (b) C. Kim, K.-I. Lim and C.-G. Song, J. Organomet. Chem., 2005, 690, 3278–3285 CrossRef CAS; (c) C. Kim, H. Kim and K. Park, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2155–2161 CrossRef CAS.
- (a) J. Yan, K. Liu, X. Zhang, W. Li and A. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 33–41 CrossRef CAS; (b) J. Yan, K. Liu, W. Li, H. Shi and A. Zhang, Macromolecules, 2016, 49, 510–517 CrossRef CAS.
- (a) W. Li, A. Zhang and A. D. Schlüter, Chem. Commun., 2008, 5523–5525 RSC; (b) W. Li, A. Zhang, K. Feldman, P. Walde and A. D. Schlüter, Macromolecules, 2008, 41, 3659–3667 CrossRef CAS.
- (a) J. Yan, W. Li, K. Liu, D. Wu, F. Chen, P. Wu and A. Zhang, Chem.–Asian J., 2011, 6, 3260–3269 CrossRef CAS PubMed; (b) J. Yan, X. Zhang, W. Li, X. Zhang, K. Liu, P. Wu and A. Zhang, Soft Matter, 2012, 8, 6371–6377 RSC.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- (a) L. Liu, W. Li, K. Liu, J. Yan, G. Hu and A. Zhang, Macromolecules, 2011, 44, 8614–8621 CrossRef CAS; (b) L. Liu, W. Li, J. Yan and A. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1706–1713 CrossRef CAS.
- J. F. Folmer-Andersen and J.-M. Lehn, J. Am. Chem. Soc., 2011, 133, 10966–10973 CrossRef CAS PubMed.
- As proved in our previous report, phase transition temperature of a thermoresponsive polymer remains unchanged if the added hydrophilic monomers cannot react with the polymer. For details, see ref. 13a.
- (a) Z.-C. Wen, R. Yang, H. He and Y.-B. Jiang, Chem. Commun., 2006, 106–108 RSC; (b) C. Agarwal and E. Prasad, New J. Chem., 2012, 36, 1859–1865 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08361g |
| This journal is © The Royal Society of Chemistry 2016 |