
Bioactive borophosphosilicate-polycaprolactone hybrid biomaterials via a non-aqueous sol gel process†
Abstract
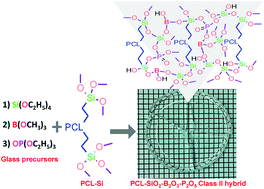
In this study, a non-aqueous sol–gel process was utilized to prepare novel class II hybrid biomaterials based on functionalized polycaprolactone (PCL) diol and borophosphosilicate glass (BPSG) as potential scaffold material for bone tissue engineering applications. PCL diol was first functionalized by reacting with (3-glycidoxypropyl)trimethoxysilane. The functionalized PCL (PCL–Si) was condensed with trimethyl borate, tetraethyl orthosilicate and triethyl phosphate via non-aqueous sol–gel reactions to form covalently bonded organic–inorganic networks. FTIR, TGA, XRD, and solid state 29Si CP-MAS NMR analyses revealed that the hybrid materials were successfully prepared. Furthermore, the hybrids were amorphous and transparent up to 60 wt% of PCL–Si content. Specifically, the organic–inorganic networks had a dominant T3 network since Si–C bond from PCL–Si is covalently bonded with the inorganic glass network and resulted in a class II hybrid. EDX and XPS studies showed uniform distribution of the various elements making up the hybrid materials. When incubated with simulated body fluids (SBF), the present hybrid materials were able to stimulate the deposition of crystalline hydroxyapatite. This study demonstrated, for the first time, the chemical reactivity of calcium-free BPSG and PCL–BPSG hybrids and their ability to deposit hydroxyapatite when incubated in SBF. The present study is also the first to incorporate B2O3 as a glass component in class II organic–inorganic hybrid biomaterials.
 Please wait while we load your content...
Please wait while we load your content...

