
Structures and electronic properties of B2Si6−/0/+: anion photoelectron spectroscopy and theoretical calculations
Guo-Jin Cao*a,
Sheng-Jie Lubc,
Hong-Guang Xubc,
Xi-Ling Xu*bc and
Wei-Jun Zhengbc
aInstitute of Molecular Science, Shanxi University, Taiyuan 030006, China. E-mail: caoguojin@sxu.edu.cn
bBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: xlxu@iccas.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 22nd June 2016
Abstract
We measured the photoelectron spectrum of B2Si6− anion and investigated the structures and electronic properties of B2Si6− anion as well as those of its neutral and cationic counterparts with quantum chemical calculations. The vertical detachment energy (VDE) of the B2Si6− anion has been measured to be 2.40 ± 0.08 eV. Through global minimum searches and CCSD(T) calculations, we have identified that the lowest-energy structures of B2Si6q (q = −1, 0, +1) are peculiar structures with a Si atom hanging over a distorted bowl-like B2Si5 framework. Quasi-planar or planar isomers have also been identified for the B2Si6 cluster at −1, 0, and +1 charge states. The quasi-planar and planar isomers are higher in energy than their bowl-like counterparts by at least 0.20 eV. The symmetries of the quasi-planar isomers varied at different charge states, ranging from Cs to C2h, then to D2h respectively for the −1, 0, and +1 charge states. The reducing of the symmetry from +1 charge state to −1 charge state is more likely due to the Jahn–Teller effect upon the addition of electrons.
1. Introduction
Silicon-based compounds have attracted tremendous attention because of their importance in the semiconductor industry.1–4 They have aromaticity, size-dependent structural characteristics, as well as distinctive physiochemical properties.5 In particular, they have been used as one of the most important p-type dopants in crystalline silicon.6,7 Boron-silicon compounds have high melting points and high hardness. Their conductivity increases with the increasing concentration of boron.5,8,9 Compared with carbon, silicon is more reluctant to form unsaturated compounds.10–14 Although the synthesis and isolation of stable unsaturated silicon compounds are a great challenge, some silicon–silicon multiple-bonded compounds were identified in the last decades. In 1981, a Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si double-bonded compound, silaethene, was isolated by Brook et al.15 In the same year, West et al.10 synthesized and isolated a stable compound containing a SiSi double bond, tetramesityldisilene. Since then, many other interesting stable unsaturated silicon compounds have been investigated extensively.16–24
Si double-bonded compound, silaethene, was isolated by Brook et al.15 In the same year, West et al.10 synthesized and isolated a stable compound containing a SiSi double bond, tetramesityldisilene. Since then, many other interesting stable unsaturated silicon compounds have been investigated extensively.16–24
Scientists have made great efforts to search for compounds with planar aromatic six-membered silicon rings (c-Si6) over the past decades because they may be used as highly stable two-dimensional (2D) aromatic nanomaterial. The cyclotrisilenylium ion25 and cyclobutadiene dianion26 have been found to be the potentially aromatic compounds based on the planar three-membered or four-membered silicon rings. As the silicon analogues of benzene, compounds with planar aromatic c-Si6 have been of great interest in the silicene field.27,28 The planar D6h c-Si6 unit in Si6H6 is less stable than the chair-like D3d structure due to the pseudo-Jahn–Teller effect.29,30 The dark green crystals of an isomer of Si6H6 with the proposed dismutational aromaticity31 were synthesized by Abersfelder et al.22 It has a chair-like conformation in line with the theoretical results.29,30 Subsequently, a cage isomer of hexasilabenzene was investigated experimentally by Abersfelder et al.32,33 and Kratzert et al.34 In addition, several planar D6h c-Si6 structures have been suggested by theoreticians. These planar benzene-like structures can be formed by substituting Si atoms by C atoms in silabenzenes.35 It has also been proposed that anionic systems c-Si62−, c-Si64−, and c-Si66− could be planar.36–38 Zdetsis et al.27 have predicted that Si6Li6 with D2h symmetry could be formed by the reduction of the c-Si66− anion in the presence of lithium. Recently, the c-BSi3 silicene containing planar aromatic D6h c-Si6 rings has been predicted by theory to be the global minimum of the BSi3 monolayer and possesses metallic character.39
There have been many experimental and theoretical investigations on the structural and electronic properties of small size SinBm clusters.8,9,39–50 Cline et al.8,9 have reported the existence of two new silicon borides SiB4 and SiB6 in silicon-boron systems. Bernardo and Morrison40 performed a theoretical study on the structures and binding energies of SiBn+ (n = 1–4) and suggested that SiBn+ (n = 1–4) cluster cations prefer planar boron networks with silicon located at the edge site. The high- and low-spin states of BSi were calculated by Boldyrev and Simons41 and were confirmed by Knight et al.42 using electron spin resonance (ESR) spectroscopy technology. Verhaegen et al.43 and Viswanathan et al.44 have studied the thermochemistry of BSi, BSi2, and BSi3. Yamauchi et al.45 have investigated the geometric and electronic properties of B12 in silicon crystalline and found that the lowest-lying isomer of B12 in silicon crystalline is icosahedral. The low-lying excited electronic states and structures of BSi2, B2Si, and B2Si2 were investigated by Davy et al. using the B3LYP method.46 Sun et al.47 have investigated BSim− (m = 1–6) cluster anions using time-of-flight mass spectrometry and quantum chemical calculations. Tam et al.50 studied the structures, thermochemical properties and growth mechanism of SinBq (n = 1–10; q = −1, 0, +1) and found that Si9B and Si10B exhibit endohedral structures. Dai et al.51 conducted a first-principle theoretical study on the structures of 2D boron-silicon compounds, and found that BnSi (n = 1–3, 5, 6) and BSim (m = 3, 4) clusters prefer planar sp2-Si structures. Recently, Dopfer and co-workers reported a combined infrared-ultraviolet two-color ionization (IR-UV2CI) spectroscopy and quantum chemical study on BSi6, and found that the most stable structure of BSi6 is a distorted pentagonal bipyramid.52
In order to get insight into the structural and electronic properties of boron-doped silicon clusters, in this work we carried out a combined photoelectron spectroscopy and quantum chemical study on B2Si6−/0/+.
2. Experimental and theoretical methods
2.1 Experimental method
The experiments were carried out on a home-built apparatus consisting of a time-of-flight mass spectrometer and a magnetic-bottle photoelectron spectrometer, which has been described elsewhere.53 The B2Si6− anion was produced by laser vaporization of rotating and translating disk targets (13 mm diameter; B/Si molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) with the second harmonic (532 nm) of a nanosecond Nd:YAG laser (Continuum Surelite II-10). Helium gas with a backing pressure of 5.0 atm. was delivered through a pulsed valve into the laser ablation source to cool the formed B2Si6− anion. The B2Si6− anion was mass-selected and then photodetached with the fourth harmonic light beam from another Nd:YAG laser (Continuum Surelite II-10, 266 nm). The resultant electrons were energy-analyzed by the magnetic-bottle photoelectron spectrometer. The photoelectron spectrum was calibrated using the spectrum of Cu− anion taken at the similar conditions. The resolution of the photoelectron spectrometer was approximately 40 meV for electrons with 1 eV kinetic energy.
:2) with the second harmonic (532 nm) of a nanosecond Nd:YAG laser (Continuum Surelite II-10). Helium gas with a backing pressure of 5.0 atm. was delivered through a pulsed valve into the laser ablation source to cool the formed B2Si6− anion. The B2Si6− anion was mass-selected and then photodetached with the fourth harmonic light beam from another Nd:YAG laser (Continuum Surelite II-10, 266 nm). The resultant electrons were energy-analyzed by the magnetic-bottle photoelectron spectrometer. The photoelectron spectrum was calibrated using the spectrum of Cu− anion taken at the similar conditions. The resolution of the photoelectron spectrometer was approximately 40 meV for electrons with 1 eV kinetic energy.
2.2 Theoretical method
The theoretical calculations of B2Si6q (q = −1, 0, +1) were performed using the Becke 3-parameter-Lee–Yang–Parr (B3LYP) density functional method as implemented in the GAUSSIAN 09 program package.54 Global minimum searches were carried out using the basin-hopping (BH) method55 at the DFT level. Low-lying structures were fully optimized using the B3LYP method with the aug-cc-pVTZ basis set. We tested the theoretical method by calculating the bond length of B–Si (1.917 Å, 4Σ−), which is in good agreement with the bond length calculated with the MP2 method in the literature (1.905 Å).42 The vibrational frequencies were calculated to confirm whether their structures are real local minima. The zero-point energies (ZPEs) corrections were included for all calculated energies. For more reliable energies, the CCSD(T) single-point calculations were performed using the Molpro2012 program56 at the B3LYP/aug-cc-pVTZ geometries. The first VDE was calculated based on the energy difference between the neutral and anion at the ground state geometry of the anion. The excited-state energies of the neutrals were calculated by using the time-dependent density functional theory (TD-DFT) method.57 The statistical average of orbital potentials (SAOP) functional was employed and all-electron basis sets TZ2P were used for B and Si. Simultaneously, the higher VDE values were also approximated using the generalized Koopman's theorem58 at the DFT/CAMY-B3LYP59 level. The TD-DFT and DFT/CAMY-B3LYP59 calculations were carried out on these clusters using Amsterdam Density Functional program (ADF 2013.01).60–62The natural bond orbital (NBO 5.0)63,64 analyses of B2Si6q (q = −1, 0, +1) were also performed to gain insights into the natural electron configurations of the valence orbitals and natural charges of these clusters. To further understand the nature of B–Si bonds in these boron-silicon complexes, we analyzed the density of state and adaptive natural density partitioning (AdNDP65) using the Multiwfn 2.6.1 package.66
3. Experimental results
The photoelectron spectrum of B2Si6− anion obtained with 266 nm photons is shown in Fig. 1. The spectral features are labeled with letters (X, A, B, and C), which correspond to the transitions from the ground state of B2Si6− anion to the ground and excited states of B2Si6 neutral. The adiabatic detachment energy (ADE) and vertical detachment energies (VDEs) estimated from the photoelectron spectrum are summarized in Table 1. The spectrum of B2Si6− anion is characterized by a small peak centered at 2.40 eV, a small shoulder centered at 3.61 eV, a broad feature centered at 3.90 eV, followed by a sharp peak centered at 4.27 eV. Because the instrumental resolution shows a significant influence on the broadening of the photoelectron spectrum, the ADE was determined by adding the instrumental resolution to the onset of the first peak in the spectrum. The onset of the first peak was determined by drawing a straight line along the leading edge of that peak across the baseline of the spectrum. The ADE of B2Si6− anion estimated from the experimental spectrum is 2.15 eV.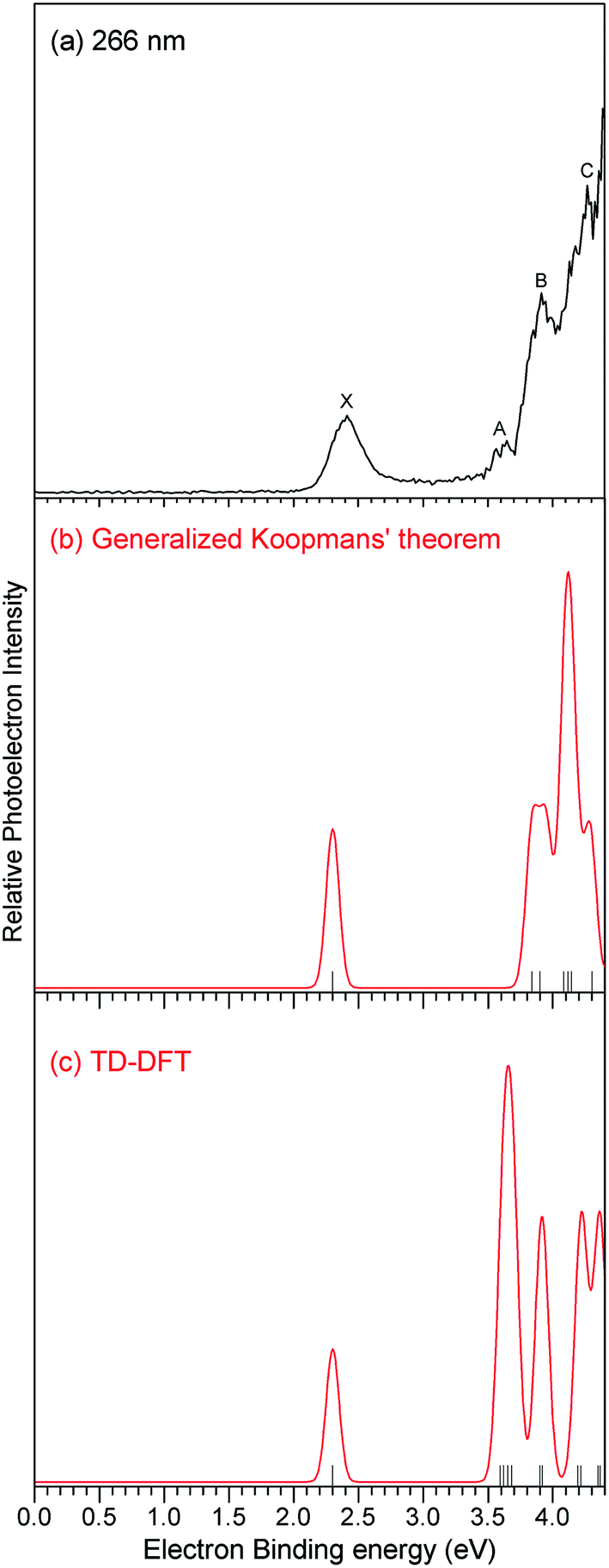 | ||
| Fig. 1 (a) Photoelectron spectrum of B2Si6− anion recorded with 266 nm photons. (b) Simulated spectrum of B2Si6− anion using the generalized Koopmans' theorem. (c) Simulated spectrum of B2Si6− anion using the TD-DFT method. The simulations were conducted by fitting the distribution of the VDE values with unit-area Gaussian functions of 0.1 eV full width at half maximum. | ||
4. Theoretical results
The structures of the typical low-lying isomers of the B2Si6− anion are shown in Fig. 2, and those of their corresponding neutral and cationic clusters are also shown in Fig. 2. The simulated spectra of B2Si6− anion using the generalized Koopmans' theorem and TD-DFT method were constructed by fitting the distribution of the VDE values with unit-area Gaussian functions of 0.10 eV full width at half maximum (Fig. 1). The relative energies, VDEs, ADEs, term energies, and NPA charges of these isomers from our calculations are summarized in Tables 2–5. | ||
| Fig. 2 Structures and relative energies of the low-lying isomers of B2Si6q (q = −1, 0, +1). The bond distances are in angstroms. The relative energies outside the parentheses are from the DFT/B3LYP method, and those in parentheses from the CCSD(T) method. | ||
| Isomers | Sym. | State | ΔE (eV) | ADE (eV) | VDE (eV) | ||||
|---|---|---|---|---|---|---|---|---|---|
| B3LYP | CCSD(T) | Theo. | Expt. | Theo. | Expt. | ||||
| B2Si6− | 1a | C1 | 2A | 0.00 | 0.00 | 2.00 | 2.15 | 2.30 | 2.40 |
| 1b | C2h | 2Bu | 0.41 | 0.31 | 2.88 | 2.98 | |||
| 1c | Cs | 2A′ | 0.51 | 0.63 | 2.16 | 2.52 | |||
| Term energies | Expt. (eV) | GKT (eV) | TD-DFT (eV) |
|---|---|---|---|
| X ← A | 1.21 | 1.59 | 1.36 |
| X ← B | 1.50 | 1.82 | 1.62 |
| X ← C | 1.87 | 1.97 | 1.92 |
| Isomers | Sym. | State | ΔE (eV) | ||
|---|---|---|---|---|---|
| B3LYP | CCSD(T) | ||||
| B2Si6 | 2a | C1 | 1A | 0.00 | 0.00 |
| 2b | C2h | 1Ag | 0.66 | 0.33 | |
| 2c | C2h | 1Ag | 1.30 | 0.45 | |
| B2Si6+ | 3a | C1 | 2A | 0.00 | 0.00 |
| 3b | D2h | 2B1g | 0.00 | 0.20 | |
| 3c | C2h | 2Au | 0.76 | 0.93 | |
| NPA charges | Spin density distributions | |||||||
|---|---|---|---|---|---|---|---|---|
| B2Si6+ (3a, C1) | B2Si6+ (3b, D2h) | B2Si6 (2a, C1) | B2Si6 (2b, C2h) | B2Si6− (1a, C1) | B2Si6+ (3a, C1) | B2Si6+ (3b, D2h) | B2Si6− (1a, C1) | |
| B1 | −0.62 | −0.77 | −1.04 | −0.73 | −1.02 | 0.00 | −0.05 | 0.00 |
| B2 | −0.48 | −0.77 | −1.04 | −0.73 | −1.07 | 0.15 | −0.05 | 0.08 |
| Si3 | 0.46 | 0.36 | 0.52 | 0.26 | 0.40 | 0.21 | 0.06 | 0.00 |
| Si4 | 0.34 | 0.36 | 0.37 | 0.26 | 0.07 | 0.08 | 0.06 | 0.11 |
| Si5 | 0.47 | 0.55 | 0.26 | 0.21 | 0.16 | 0.45 | 0.43 | 0.00 |
| Si6 | 0.28 | 0.36 | 0.31 | 0.26 | 0.26 | 0.04 | 0.06 | 0.00 |
| Si7 | 0.39 | 0.36 | 0.57 | 0.26 | 0.17 | 0.00 | 0.06 | 0.73 |
| Si8 | 0.16 | 0.55 | 0.05 | 0.21 | 0.03 | 0.07 | 0.43 | 0.08 |
4.1 B2Si6−
For the B2Si6− anion, the most stable isomer 1a can be viewed as one Si atom located on the top of the distorted bowl-like B2Si5 framework. The distance between B1 and Si8 is ∼2.274 Å. The Si3–Si4, Si4–Si5, Si5–Si6 and Si6–Si7 bond lengths are ∼2.455 Å. The B–B bond distance is ∼1.682 Å, close to the B–B bond lengths (∼1.710 Å) in the compounds of the a-B (a-rhombohedral boron) type.67 The first VDE and ADE of isomer 1a are calculated to be 2.30 eV and 2.00 eV at the DFT/B3LYP level, respectively, in reasonable agreement with the experimental values (2.40 eV and 2.15 eV). The first term energies ΔE (B2Si6(X) ← B2Si6(A)) obtained from the generalized Koopmans' theorem and TD-DFT calculations are 1.59 eV and 1.36 eV, respectively, in reasonable agreement with the experimental value (1.21 eV). The second term energies ΔE (B2Si6(X) ← B2Si6(B)) obtained from the generalized Koopmans' theorem and TD-DFT calculations are 1.82 eV and 1.62 eV, respectively, also in reasonable agreement with the experimental measurement (1.50 eV). The second detachment channels correspond to the transitions to the two 3A final states with electrons detached from HOMO−1 and HOMO−2 of B2Si6− anion. The third term energies ΔE (B2Si6(X) ← B2Si6(C)) obtained from the generalized Koopmans' theorem and TD-DFT calculations are 1.97 eV and 1.92 eV, respectively, also in good agreement with the experimental value (1.87 eV). The simulated spectra of the bowl-like isomer 1a from the generalized Koopmans' theorem and TD-DFT calculations can reasonably explain all the observed photoelectron spectrum bands (Fig. 1). Isomer 1b is a C2h distorted oblique prism with the 2Bu electronic state. The theoretical VDE of isomer 1b is calculated to be 2.98 eV, which does not match with the experimental value. Isomer 1c is a quasi-planar structure with a boron dimer surrounded by six Si atoms. It is of Cs symmetry with the 2A′ electronic state. The VDE and ADE of isomer 1c are calculated to be 2.52 eV and 2.16 eV, respectively. Isomers 1b and 1c are less stable than isomer 1a by 0.41 eV and 0.51 eV at the DFT/B3LYP level, respectively. The CCSD(T)/aug-cc-pVTZ results show that isomers 1b and 1c are higher in energy than isomer 1a by 0.31 eV and 0.63 eV, respectively. Therefore, the existence of isomers 1b and 1c in the experiments can be ruled out, and isomer 1a corresponds to the major peaks observed in the spectrum of the B2Si6− anion.4.2 B2Si6
The lowest-energy structure of B2Si6 (isomer 2a) with no symmetry is similar to isomer 1a of B2Si6− anion. The Si3–Si4, Si4–Si5, Si5–Si6, and Si6–Si7 bond lengths are also ∼2.455 Å. The B–B bond distance is 1.637 Å, close to the B–B bond length of isomer 1a. But the Si7–Si8 bond length is ∼2.795 Å in isomer 2a, much shorter than that (∼3.820 Å) of isomer 1a. Isomer 2b is of C2h symmetry in 1Ag electronic state with a boron dimer surrounded by six Si atoms. It is less stable in energy than isomer 2a by 0.66 eV from the B3LYP/aug-cc-pVTZ calculations, while CCSD(T) calculations show that the energy difference is 0.33 eV. Isomer 2c also has a C2h symmetry with the 1Ag electronic state and it is higher in energy than isomer 2a by 1.30 eV and 0.45 eV from the B3LYP and CCSD(T) calculations, respectively. It is worth mentioning that we also considered the highly symmetric D2h structure of isomer 2b, however, it is unstable.To understand the stabilities of the isomers 2a and 2b of B2Si6, we searched the possible transition barriers between them by Berny method. The transition-state structure obtained by us was confirmed to connect the correct reactants (isomer 2a) and products (isomer 2b) by intrinstic reaction coordinate (IRC). The transition state is determined for the conversion pathway, with a conversion barrier of 0.48 eV calculated at the CCSD(T) level. It indicates that the bowl-like structure of B2Si6 neutral (isomer 2a) is highly stable.
4.3 B2Si6+
The low-lying isomers of B2Si6+ cation (3a and 3b) are degenerated in energy from the B3LYP results. However, the CCSD(T) results show that isomer 3b is higher than isomer 3a by 0.20 eV in energy. The structure of isomer 3a is similar to isomer 2a of B2Si6. The Si7–Si8 and Si4–Si8 bond lengths are slightly elongated in isomer 3a relative to those of isomer 2a. Isomer 3b has a perfectly planar hexagon structure with a large HOMO–LUMO gap of 2.35 eV. It has a D2h symmetry with the 2B1g electronic state. The Si–Si distances are somewhat different in isomer 3b. The Si4–Si5, Si5–Si6, Si6–Si7, and Si3–Si4 bond distances are ∼2.500 Å, slightly shorter than the Si7–Si8 and Si4–Si8 bonds (∼2.900 Å) in the same isomer. The B–B bond length in isomer 3b is ∼1.666 Å, close to the B–B distances in the quasi-planar isomers of the negative and neutral charge states. Isomer 3c has a C2h symmetry with the 2Au electronic state and it is higher in energy than isomer 3a by 0.76 eV and 0.93 eV from the B3LYP and CCSD(T) calculations, respectively, indicating that the preference of isomer 3a is the largest. It is worth mentioning that, for the quasi-planar or planar structures, the symmetries of the negatively charged B2Si6− (1c, Cs) and neutral B2Si6 (2b, C2h) are lower than that of the positively charged B2Si6+ (3b, D2h), indicating that the Jahn–Teller effect caused by the increased electrons might be the major factor contributing to the lowing of the symmetry.5. Discussion
5.1 NPA charges and spin density distributions of B2Si6q (q = −1, 0, +1)
As shown in Table 5, the effective atomic charges of the most stable structures of B2Si6q (q = −1, 0, +1) indicate that both B atoms in the clusters are negatively charged, QB < 0. It is obvious that some electrons are transferred to the B atoms, partly because of the larger Pauling electronegativity of B (2.04) as compared to Si (1.90).68 The electron is mainly detached from the p orbitals of Si(4) and lateral Si(7) atoms when B2Si6− anion with a C1 symmetry was photodetached. Inversely, the electron is taken off from the B atoms when the lowest-energy isomer of B2Si6 neutral loses one electron. The spin density distributions of B2Si6− anion (1a, C1) have shown that its spin densities are mainly located on lateral Si(7) and slightly located on B(2), Si(4), and Si(8). For B2Si6+ cation (3a, C1), the spin densities are mainly located on B(2), Si(3), and Si(5). For planar B2Si6+ cation (3b, D2h), the unpaired electron is mainly located on the C2 axis of Si(5) and Si(8).5.2 Molecular orbitals of B2Si6 (2a, C1)
Using the ‘adaptive density partitioning’ approach,65 one can also approximately extract 9 delocalized valence MOs of approximate “s” and “p” symmetry for the remaining 30 electrons in the B2(2s22p1)–Si6(3s23p2) valence shell of B2Si6 (2a, C1) (Fig. 3), in addition to the 6 lone pair electrons of six Si atoms, 9 delocalized MOs include one 2c-2e BSi σ bond (electronic occupation is equal to 1.88 |e|), three 3c-2e B2Si or BSi2 σ bonds (electronic occupation is equal to 1.89 |e|), three 4c-2e BSi3 σ bonds (electronic occupation is equal to 1.90 |e|), and two 5c-2e B2Si3 MOs (electronic occupation is equal to 1.91 |e|). The 15 electron pairs of these 30e systems cannot be localized symmetrically. The two 5c-2e B2Si3 MOs contain the σ plus π double delocalization. The Si7–Si8 bond length in isomer 2a is shortened by 1.025 Å relative to that of isomer 1a, most likely due to the forming of the 4c-2e BSi3 σ bonds. | ||
| Fig. 3 Molecular orbitals of the bowl-like isomer of B2Si6 (2a, C1). Six lone pairs of Si atoms and nine optimally localized MOs (contour values ψ = ±0.038 au) are from the AdNDP65 method (ON = electronic occupation). | ||
6. Conclusions
We measured the photoelectron spectrum of B2Si6− anion and investigated its anionic, cationic and neutral structures with density functional theory (B3LYP) and wave function theory (CCSD(T)) approaches. By comparing the calculated VDEs with the experimental measurements, the structure of B2Si6− anion is determined. The CCSD(T) results reveal that the lowest-lying isomers of B2Si6q (q = −1, 0, +1) have a peculiar structure with a Si atom hanging over a distorted bowl-like B2Si5 framework. The Si7–Si8 distance in the lowest-lying isomer 2a of B2Si6 neutral is shortened by 1.025 Å relative to that of the lowest-lying isomer 1a of B2Si6− anion. Isomer 2a is characterized with σ or π delocalization in chemical bonding. In addition to the bowl-like structures, we also found low-lying quasi-planar or planar structures for B2Si6q (q = −1, 0, +1), which are higher in energy than the corresponding bowl-like structures by at least 0.20 eV. The planar B2Si6+ cation has a D2h symmetry and is highly aromatic. For the quasi-planar or planar structures, the symmetries of the negatively charged B2Si6− (1c, Cs) and neutral B2Si6 (2b, C2h) are lower than that of the positively charged B2Si6+ (3b, D2h), more likely due to the Jahn–Teller effect upon the addition of electrons.Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No. 21103202 and 21501114) and Natural Science Foundation of Shanxi Province (Grant No. 2015021048).Notes and references
- H. Hiura, T. Miyazaki and T. Kanayama, Phys. Rev. Lett., 2001, 86, 1733–1736 CrossRef CAS PubMed.
- S. N. Khanna, B. K. Rao and P. Jena, Phys. Rev. Lett., 2002, 89, 016803 CrossRef CAS PubMed.
- A. Fleurence, R. Friedlein, T. Ozaki, H. Kawai, Y. Wang and Y. Yamada-Takamura, Phys. Rev. Lett., 2012, 108, 245501 CrossRef PubMed.
- L. Chen, C.-C. Liu, B. Feng, X. He, P. Cheng, Z. Ding, S. Meng, Y. Yao and K. Wu, Phys. Rev. Lett., 2012, 109, 056804 CrossRef PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. Lett., 2003, 90, 055502 CrossRef PubMed.
- Y. Cui, X. Duan, J. Hu and C. M. Lieber, J. Phys. Chem. B, 2000, 104, 5213–5216 CrossRef CAS.
- G. Cantele, E. Degoli, E. Luppi, R. Magri, D. Ninno, G. Iadonisi and S. Ossicini, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 113303 CrossRef.
- C. F. Cline, Nature, 1958, 181, 476–477 CrossRef CAS.
- C. F. Cline and D. E. Sands, Nature, 1960, 185, 456 CrossRef CAS.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- S. Ishida, T. Iwamoto, C. Kabuto and M. Kira, Nature, 2003, 421, 725–727 CrossRef CAS PubMed.
- A. Sekiguchi and V. Y. Lee, Chem. Rev., 2003, 103, 1429–1448 CrossRef CAS PubMed.
- M. Weidenbruch, Angew. Chem., Int. Ed., 2003, 42, 2222–2224 CrossRef CAS PubMed.
- N. Tokitoh, Acc. Chem. Res., 2004, 37, 86–94 CrossRef CAS PubMed.
- A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst and R. K. Kallury, J. Chem. Soc., Chem. Commun., 1981, 191–192 RSC.
- A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755–1757 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed.
- H. Ottosson and P. G. Steel, Chem.–Eur. J., 2006, 12, 1576–1585 CrossRef CAS PubMed.
- M. Weidenbruch, Angew. Chem., Int. Ed., 2006, 45, 4241–4242 CrossRef CAS PubMed.
- M. Kira and T. Iwamoto, Adv. Organomet. Chem., 2006, 54, 73–148 CrossRef CAS.
- T. Sasamori, J. S. Han, K. Hironaka, N. Takagi, S. Nagase and N. Tokitoh, Pure Appl. Chem., 2010, 82, 603–612 CrossRef CAS.
- K. Abersfelder, A. J. White, H. S. Rzepa and D. Scheschkewitz, Science, 2010, 327, 564–566 CrossRef CAS PubMed.
- T. Agou, Y. Sugiyama, T. Sasamori, H. Sakai, Y. Furukawa, N. Takagi, J.-D. Guo, S. Nagase, D. Hashizume and N. Tokitoh, J. Am. Chem. Soc., 2012, 134, 4120–4123 CrossRef CAS PubMed.
- T. Sasamori, K. Hironaka, Y. Sugiyama, N. Takagi, S. Nagase, Y. Hosoi, Y. Furukawa and N. Tokitoh, J. Am. Chem. Soc., 2008, 130, 13856–13857 CrossRef CAS PubMed.
- M. Ichinohe, M. Igarashi, K. Sanuki and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 9978–9979 CrossRef CAS PubMed.
- V. Y. Lee, K. Takanashi, T. Matsuno, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2004, 126, 4758–4759 CrossRef CAS PubMed.
- A. D. Zdetsis, J. Chem. Phys., 2007, 127, 214306 CrossRef PubMed.
- D. Jose and A. Datta, J. Phys. Chem. C, 2012, 116, 24639–24648 CrossRef CAS.
- S. Nagase, H. Teramae and T. Kudo, J. Chem. Phys., 1987, 86, 4513–4517 CrossRef CAS.
- K. K. Baldridge, O. Uzan and J. M. Martin, Organometallics, 2000, 19, 1477–1487 CrossRef CAS.
- C. Gerdes and T. Müller, Angew. Chem., Int. Ed., 2010, 49, 4860–4862 CrossRef CAS PubMed.
- K. Abersfelder, A. J. White, R. J. Berger, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2011, 123, 8082–8086 CrossRef.
- K. Abersfelder, A. J. White, R. J. Berger, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2011, 50, 7936–7939 CrossRef CAS PubMed.
- D. Kratzert, D. Leusser, J. J. Holstein, B. Dittrich, K. Abersfelder, D. Scheschkewitz and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 4478–4482 CrossRef CAS PubMed.
- A. S. Ivanov and A. I. Boldyrev, J. Phys. Chem. A, 2012, 116, 9591–9598 CrossRef CAS PubMed.
- M. Takahashi and Y. Kawazoe, Organometallics, 2005, 24, 2433–2440 CrossRef CAS.
- M. Takahashi and Y. Kawazoe, Chem. Phys. Lett., 2006, 418, 475–480 CrossRef CAS.
- M. Takahashi and Y. Kawazoe, Comput. Mater. Sci., 2006, 36, 30–35 CrossRef CAS.
- X. Tan, F. Li and Z. Chen, J. Phys. Chem. C, 2014, 118, 25825–25835 CrossRef CAS.
- D. N. Bernardo and G. H. Morrison, Surf. Sci., 1989, 223, L913–L919 CrossRef CAS.
- A. I. Boldyrev and J. Simons, J. Phys. Chem., 1993, 97, 1526–1532 CrossRef CAS.
- J. L. B. Knight, A. J. McKinley, R. M. Babb, M. D. Morse and C. A. Arrington, J. Chem. Phys., 1993, 98, 6749–6757 CrossRef.
- G. Verhaegen, F. E. Stafford and J. Drowart, J. Chem. Phys., 1964, 40, 1622–1628 CrossRef CAS.
- R. Viswanathan, R. W. Schmude and K. A. Gingerich, J. Phys. Chem., 1996, 100, 10784–10786 CrossRef CAS.
- J. Yamauchi, N. Aoki and I. Mizushima, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, R10245 CrossRef CAS.
- R. Davy, E. Skoumbourdis and D. Dinsmore, Mol. Phys., 2005, 103, 611–619 CrossRef CAS.
- Z. Sun, Z. Yang, Z. Gao and Z. Tang, Rapid Commun. Mass Spectrom., 2007, 21, 792–798 CrossRef CAS.
- E. Bourgeois and X. Blase, Appl. Phys. Lett., 2007, 90, 142511 CrossRef.
- X. Pi, X. Chen and D. Yang, J. Phys. Chem. C, 2011, 115, 9838–9843 CrossRef CAS.
- N. M. Tam, T. B. Tai and M. T. Nguyen, J. Phys. Chem. C, 2012, 116, 20086–20098 CrossRef CAS.
- J. Dai, Y. Zhao, X. J. Wu, J. L. Yang and X. C. Zeng, J. Phys. Chem. Lett., 2013, 4, 561–567 CrossRef CAS PubMed.
- N. X. Truong, M. Haertelt, B. K. Jaeger, S. Gewinner, W. Schöllkopf, A. Fielicke and O. Dopfer, Int. J. Mass Spectrom., 2016, 395, 1–6 CrossRef CAS.
- H.-G. Xu, Z.-G. Zhang, Y. Feng, J. Yuan, Y.-C. Zhao and W.-J. Zheng, Chem. Phys. Lett., 2010, 487, 204–208 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- H.-J. Zhai, Y.-F. Zhao, W.-L. Li, Q. Chen, H. Bai, H.-S. Hu, Z. A. Piazza, W.-J. Tian, H.-G. Lu and Y.-B. Wu, Nat. Chem., 2014, 6, 727–731 CrossRef CAS PubMed.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, et al., MOLPRO, Version 2012.1, A Package of ab Initio Programs, see http://www.molpro.net Search PubMed.
- B. Sengupta, C. M. Ritchie, J. G. Buckman, K. R. Johnsen, P. M. Goodwin and J. T. Petty, J. Phys. Chem. C, 2008, 112, 18776–18782 CrossRef CAS.
- K. Gillen, R. Jensen and N. Davidson, J. Am. Chem. Soc., 1964, 86, 2792–2796 CrossRef CAS.
- M. Seth and T. Ziegler, J. Chem. Theory Comput., 2012, 8, 901–907 CrossRef CAS PubMed.
- C. F. Guerra, J. Snijders, G. Te Velde and E. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- See http://www.scm.com for ADF2012.01 and ADF2013.01, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- B. Albert and H. Hillebrecht, Angew. Chem., Int. Ed., 2009, 48, 8640–8668 CrossRef CAS PubMed.
- L. C. Allen, J. Am. Chem. Soc., 1989, 111, 9003–9014 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |