
Cross-linkable polymers containing a triple bond backbone and their application in photovoltaic devices
Thi Thu Trang Buiab,
Sangheon Parkad,
Muhammad Jahandarac,
Chang Eun Songa,
Sang Kyu Leeac,
Jong-Cheol Leeac,
Sang-Jin Moonac and
Won Suk Shin*ac
aEnergy Materials Research Center, Korea Research Institute of Chemical Technology (KRICT), 141 Gajeongro, Yuseong, Daejeon, 305-600, Korea
bFaculty of Environment, Vietnam National University of Agriculture, Hanoi, Vietnam
cDepartment of Nanomaterials Science and Engineering, University of Science and Technology (UST), 217 Gajeongro, Yuseong, Daejeon, 305-350, Korea. E-mail: shinws@krict.re.kr
dDepartment of Physics, Sungkyunkwan University (SKKU), 2066 Seoburo, Jangan, Suwon, Gyeong Gi-do, Korea
First published on 10th June 2016
Abstract
Two novel polymers containing triple bonds in the backbone with different conjugation types (acceptor–acceptor and acceptor–donor structures) were synthesized and investigated for their crosslinking characteristics under UV irradiation. The crosslink formation was proven via UV-vis and IR spectroscopy, and it was found that the crosslinked polymers have solvent resistance properties during subsequent solvent washing with a similar solvent for the active layer. The two triple bond polymers were used as buffer layer materials to modify the interface properties of electron-selective ZnO in inverted PSCs via a spin-coating process. The effects of the buffer layer on the surface of the ZnO were studied via atomic force microscopy and contact angle measurements. The increased hydrophobic nature of the ZnO surface resulted in better contact with the active layer, and led to an improvement in the performance of the photovoltaic devices with increased fill factor (FF).
1. Introduction
Because of their many advantages, such as being lightweight, low cost, having flexible substrates etc., bulk heterojunction (BHJ) organic solar cells (OSCs) have attracted considerable attention recently. During the past decade, their power conversion efficiency (PCE) has been increased significantly and has reached over 11% by the use of novel polymers and control of the active layer morphology.1,2 In general, the fabrication of multilayer organic electronic devices including OSCs is carried out by one of two methods: high-vacuum vapor deposition or solution processing. High vacuum vapor deposition can be used for most small molecule-based devices but is relatively expensive and time consuming, and it is difficult to make large-area devices and apply to high-molecular weight materials.3 In contrast, solution processes have the potential to facilitate rapid and low-cost processing, fabrication on a large size scale, and can be used for all soluble materials. However, during multilayer integration, the deposition of a second layer from solution can lead to partial dissolving of the previous layer if the solvent for the second layer materials also dissolves the first layer materials.4 This is the big challenge for the solution processing method to fabricate multilayer devices compared to the high-vacuum vapor deposition method. To overcome this, a number of efforts have been explored. One of the approaches is the development of novel materials that can provide excellent solvent resistance after thermo- or photo-crosslinking or chemical treatments.5,6 These cross-linkable materials can be used as electrode buffer layer materials7,8 to modify the interfacial properties of the charge transporting layer of organic electronic devices.In the past, polymers containing triple bonds in the pendant or backbone were found to easily undergo crosslinking on exposure to ultraviolet light and the crosslinking reaction could also be sensitized by additives.9,10 The formation of only a few fractions of crosslinks is probably sufficient to insolubilize the polymer. So, triple bond containing polymers are one of the potential candidates as crosslinkable materials which can be used for fabricating multilayer devices by solution processing. In this regard, we firstly tried the synthesis of a series of polymers containing triple bonds with different conjugated backbone structures: donor–acceptor (D–A), donor–donor (D–D), and acceptor–acceptor (A–A). Two polymers, D–A and A–A, were tested for their crosslinkability under UV irradiation. We expected that the crosslinked layer would not be washed out when the next layer was introduced. These polymers were then used as buffer layers on top of ZnO to improve the performance of inverted polymer solar cells (PSCs).
2. Experimental section
2.1. Instruments and characterization
Molecular weights were determined using GPC on a Viscotek TSA302 Triplet Detector Array system in CHCl3 using a polystyrene standard at room temperature. Absorption spectra were recorded using a Shimadzu UV-3600 UV-visible spectrometer. Samples for UV-vis absorption measurements were prepared by spin-coating the solution of polymer in chloroform on cleaned quartz glass substrates. CV was measured by using an IviumStat instrument. CV was conducted with a scan rate of 50 mV s−1 at room temperature under the protection of argon with 0.1 M tetrabutylammonium tetrafluoroborate in acetonitrile as the electrolyte. A platinum electrode was coated with a thin copolymer film and used as the working electrode. A Pt wire was used as the counter electrode, and a Ag/AgNO3 (0.1 M) electrode was used as the reference electrode.2.2. Fabrication and characterization of polymer solar cells
The structure of the BHJ device is ITO/ZnO/Interlayer polymer/P3HT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC61BM (1.0:0.8 w/w)/MoOx/Al. To fabricate the PSCs, first, ITO coated glass slides were cleaned with detergent, followed by ultrasonic washing in D.I. water, acetone and IPA sequentially, and dried in an oven overnight. After UV-ozone treatment for 10 min, a ZnO solution was spin-coated onto the ITO substrate at 6000 rpm for 40 s and then annealed at 200 °C for 1 hour. For deposition of the active layer, P3HT:PC61BM (1.0:0.8 w/w) dissolved in o-DCB was spin-cast on top of the ZnO layer in a nitrogen-filled glove box. Finally, the metal top electrode, MoOx and Ag, was sequentially deposited onto the BHJ active layer under vacuum (<2 × 10−6 Torr) by thermal evaporation.
:PC61BM (1.0:0.8 w/w)/MoOx/Al. To fabricate the PSCs, first, ITO coated glass slides were cleaned with detergent, followed by ultrasonic washing in D.I. water, acetone and IPA sequentially, and dried in an oven overnight. After UV-ozone treatment for 10 min, a ZnO solution was spin-coated onto the ITO substrate at 6000 rpm for 40 s and then annealed at 200 °C for 1 hour. For deposition of the active layer, P3HT:PC61BM (1.0:0.8 w/w) dissolved in o-DCB was spin-cast on top of the ZnO layer in a nitrogen-filled glove box. Finally, the metal top electrode, MoOx and Ag, was sequentially deposited onto the BHJ active layer under vacuum (<2 × 10−6 Torr) by thermal evaporation.
The J–V characteristics of the devices were recorded using a solar simulator with a Keithley 2400 source measure unit. Characterization of the un-encapsulated solar cells was carried out in air under the illumination of AM 1.5G, 100 mW cm−2, using a solar simulator (McScience, Inc.) with a xenon light source. The illumination intensity was set using an NREL certified silicon diode with an integrated KG5. The external quantum efficiency (EQE) was measured using a reflective microscope objective to focus the light output from a 100 W halogen lamp fitted with a monochrometer and an optical chopper (McScience, Inc.).
2.3. Materials
2,7-Dibromo-9,9-didecylfluorene,11 4,7-bisethynyl-2,1,3-benzothiadiazole,12 2,5-bis(5-bromo-3-hexylthiophene-2-yl)thiazolo[5,4-d]thiazole,13 and N-heptadecan-9′-yl-2,7-dibromocarbazole14 were synthesized according to the procedures reported in the literature. Other reagents and solvents were purchased from Sigma Aldrich, Tokyo Chemical Industry Co., Ltd and 4Chem Laboratory. All chemicals were used as received. Other monomers were synthesized according to Scheme 1.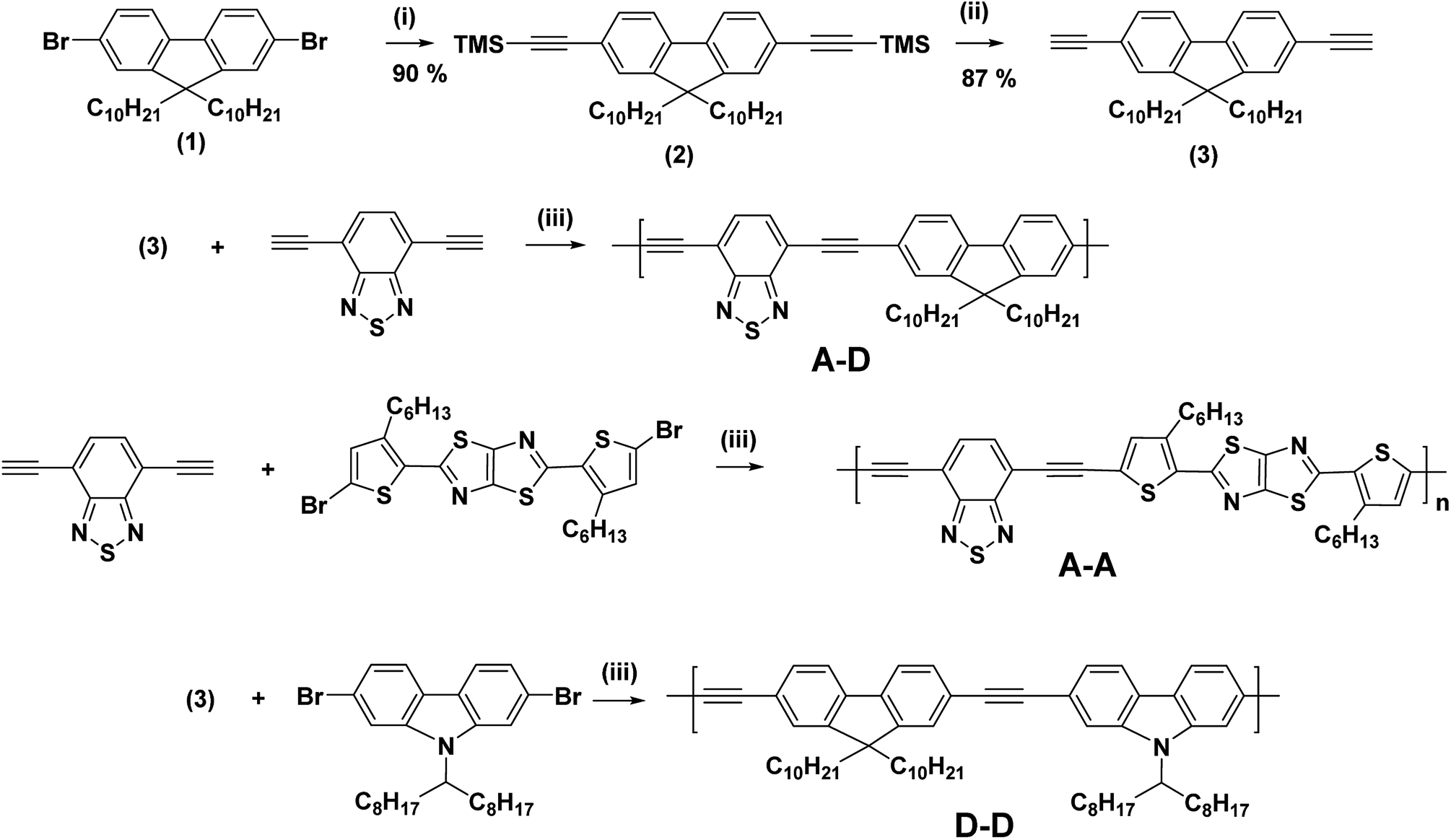 | ||
| Scheme 1 Synthetic routes of monomers and polymers. | ||
A mixture of 2,7-dibromo-9,9-didecylfluorene (12.090 g, 20 mmol), tetrakis (triphenylphosphine palladium (0)) (0.464 g, 0.4 mmol), copper(I) iodide (0.152 g, 0.8 mmol) and trimethylsilyl acetylene (5.893 g, 60.00 mmol) was dissolved in 140 mL of triethylamine. The reaction mixture was stirred at 75 °C overnight under an Ar atmosphere. After cooling to room temperature, the reaction solvent was removed under reduced pressure. The residue was extracted with CH2Cl2. The organic layer was washed with cool water and aqueous 1.2 N HCl then dried over anhydrous MgSO4, and finally evaporated under reduced pressure. The crude was purified by silica gel column chromatography eluting with CH2Cl2/hexane (1
 :4) to obtain 2,7-bis(2-trimethylsilyl)ethynyl-9,9-didecylfluorene (12.66 g, 99%) as a yellow oil.
:4) to obtain 2,7-bis(2-trimethylsilyl)ethynyl-9,9-didecylfluorene (12.66 g, 99%) as a yellow oil.1H-NMR (300 MHz, CDCl3, δ ppm): 7.60 (d, 2H), 7.46 (dd, 2H), 7.42 (s, 2), 1.77–1.71 (m, 4H), 1.12–0.83 (m, 32H), 0.72–0.64 (t, 6H), 0.40 (s, 18H).
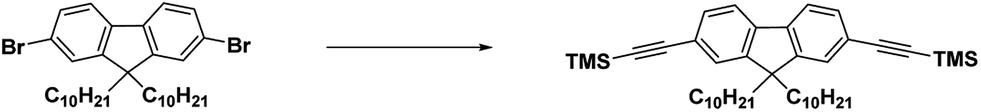
A mixture of 2,7-bis(2-trimethylsilyl)ethynyl-9,9-didecylfluorene (12.660 g, 19.80 mmol) in 500 mL of 1 mol L−1 KOH methanol solution was stirred at room temperature in the dark for 1 hour. The reaction mixture was poured into cool water (1000 mL) and extracted with CH2Cl2 several times. The combined organic layers were dried over anhydrous MgSO4 and then the solvent was removed. The residue was further purified using silica gel column chromatography (with hexane as the eluent) to afford 2,7-bisethynyl-9,9-didecylfluorene as a yellow oil (8.56 g, 87%). This compound was stored in the dark under nitrogen at 10 °C.
1H-NMR (300 MHz, CDCl3, δ ppm): 7.60 (d, 2H), 7.45 (d, 2H), 7.41 (s, 2), 3.06 (s, 2H), 1.77–1.71 (m, 4H), 1.12–0.89 (m, 32H), 0.72–0.64 (t, 6H).
2,7-Dibromo-9,9-didecylfluorene (241.1 mg, 0.40 mmol), 4,7-bisethynyl-2,1,3-benzothiadiazole (73.7 mg, 0.40 mmol), Pd(PPh3)4 (46 mg, 0.04 mmol) and CuI (15 mg, 0.08 mmol) were dissolved in a mixed solvent of THF (3.4 mL) and TEA (1.7 mL) under a nitrogen atmosphere. The D–A polymer was obtained as a brown solid (125 mg, 50% yield). Mw = 12.2 kg mol−1, PDI: 1.487.

2,7-Bisethynyl-9,9-didecylfluorene (203.4 mg, 0.40 mmol), N-heptadecan-9′-yl-2,7-dibromocarbazole (225.4 mg, 0.40 mmol), Pd(PPh3)4 (46 mg, 0.04 mmol) and CuI (15 mg, 0.08 mmol) were dissolve in a mixed solvent (THF (5 mL) and Et3N (1.7 mL)) under a nitrogen atmosphere. The D–D crude polymer was obtained as a yellow solid, but was insoluble in common chlorinated solvents (CHCl3, CB, o-DCB…) even under hot conditions.

2,5-Bis(5-bromo-3-hexylthiophene-2-yl)thiazolo[5,4-d]thiazole (253.0 mg, 0.40 mmol), 4,7-bisethynyl-2,1,3-benzothiadiazole (73.7 mg, 0.40 mmol), Pd(PPh3)4 (46 mg, 0.04 mmol) and CuI (15 mg, 0.08 mmol) were dissolved in a mixed solvent of THF (3.4 mL) and Et3N (1.7 mL) under a nitrogen atmosphere. The A–A polymer was obtained as a purple solid (70 mg, 27% yield). Mw = 14.0 kg mol−1, PDI: 2.576.

3. Results and discussion
3.1. Material synthesis
The synthetic routes of the monomers and corresponding polymers are illustrated in Scheme 1. The polymers were obtained by Sonogashira cross-coupling reaction between the dibromo and bisethynyl compounds in the presence of Pd(PPh3)4 and CuI as catalysts and with a solvent system of triethylamine (TEA)/THF (1:2, v/v) via thermal heating for 48 hours to obtain the D–A, D–D and A–A polymers. However, after the reaction, the D–D crude product was insoluble in chloroform so we could not purify and investigate it further. The two other crude polymer products, D–A and A–A, were precipitated in MeOH and purified by Soxhlet extraction. The pure polymers D–A and A–A collected from the chloroform fraction display high solubility in chlorinated organic solvents such as chlorobenzene (CB), o-dichlorobenzene (o-DCB), and chloroform. The yields of the polymerizations to synthesize D–A and A–A are 50% and 27%, respectively. The number-average molecular weight (Mn) values for D–A and A–A are 12.2 and 14.0 kg mol−1, with polydispersity indexes (PDIs) of 1.487 and 2.576, respectively, as determined by gel permeation chromatography (GPC) using chloroform as the eluent, calibrated with polystyrene standards.
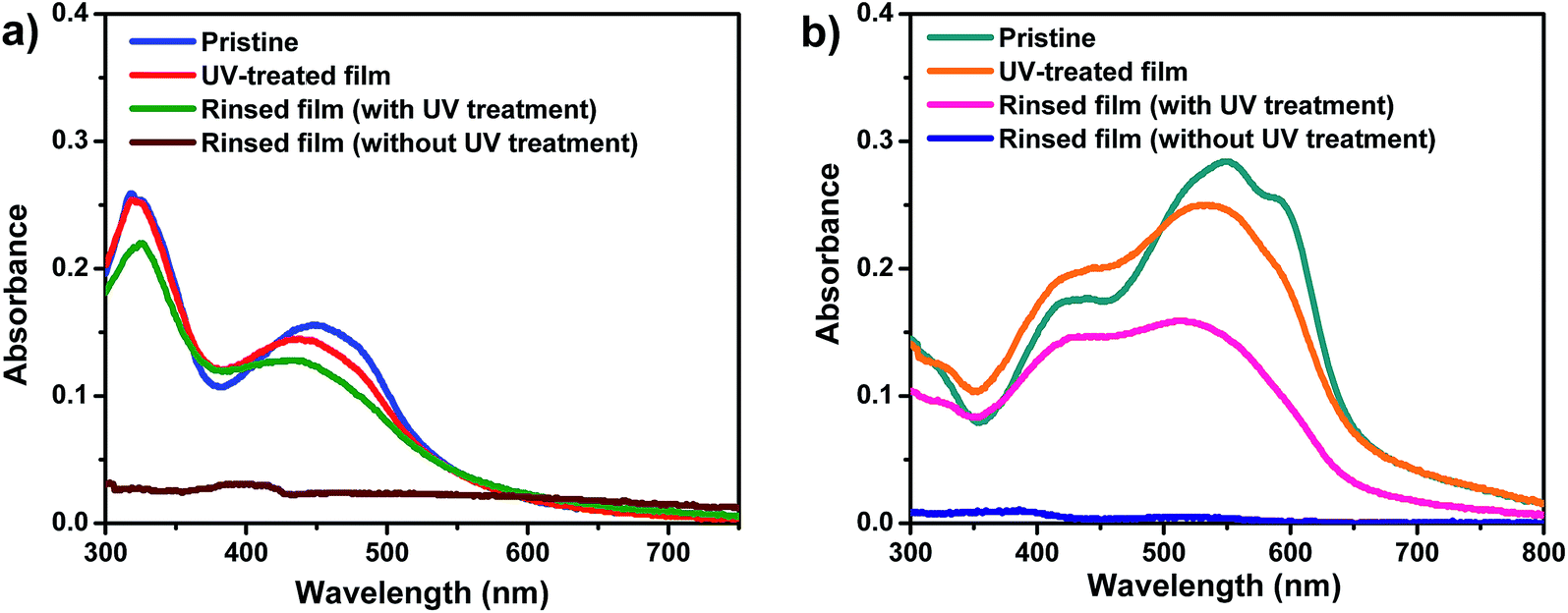
From the UV spectra of the pristine films (Fig. 1), the optical properties of the two polymers are summarized in Table 1, and the band gaps of polymers D–A and A–A were calculated to be 2.25 and 1.90 eV, respectively.
 | ||
| Fig. 1 UV-vis absorption spectra of thin films of the polymers: (a) polymer D–A, and (b) polymer A–A. | ||
3.2. Crosslinkability of the polymers
The crosslinkabilities of polymers D–A and A–A were investigated via UV-vis spectroscopy and infrared (IR) spectroscopy. Crosslinking was performed using UV-irradiation with a wavelength of 254 nm. First, the neat polymer films, prepared by spin-coating on quartz substrates, were exposed to UV light with a wavelength of 254 nm for 10 min under inert gas. After that, these treated films were analysed by UV-vis spectroscopy before and after rinsing with chloroform. Another polymer film without UV treatment was also rinsed with chloroform and the UV-vis spectrum was obtained for comparison. Fig. 1 shows the UV-vis spectra of the polymer thin films under different conditions: pristine, rinsed with chloroform, exposed to UV, and rinsed with chloroform after exposure to UV. In the case of both polymers, the absorption spectra of the pristine films are almost disappeared after rinsing with chloroform. This means that the neat thin films can be mostly washed out by the solvent used for the following spin coating. However, the films exposed to UV light maintained most of their UV-vis absorption after rinsing with chloroform. So the generation of this cross-linked network seems to be made by exposure to UV light. These cross-linked layers will allow the introduction of a subsequent active layer by spin-coating without being destroyed.To prove the cross-linking, IR spectroscopy is performed. The thin films of polymers were fabricated on the surface of KBr plates and IR spectra were obtained before and after UV exposure to see the change of vibrational stretching in the alkyne functional groups in the polymer backbone. As can be seen in Fig. 2, the vibrational stretching of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C group in the triple bond at 2191 cm−1 (A–A) and 2192 cm−1 (D–A) was significantly reduced after the UV irradiation, proving the occurrence of cross-linking. The changes of the UV spectra shape after UV exposure also support the structural change of the polymers.
C group in the triple bond at 2191 cm−1 (A–A) and 2192 cm−1 (D–A) was significantly reduced after the UV irradiation, proving the occurrence of cross-linking. The changes of the UV spectra shape after UV exposure also support the structural change of the polymers.
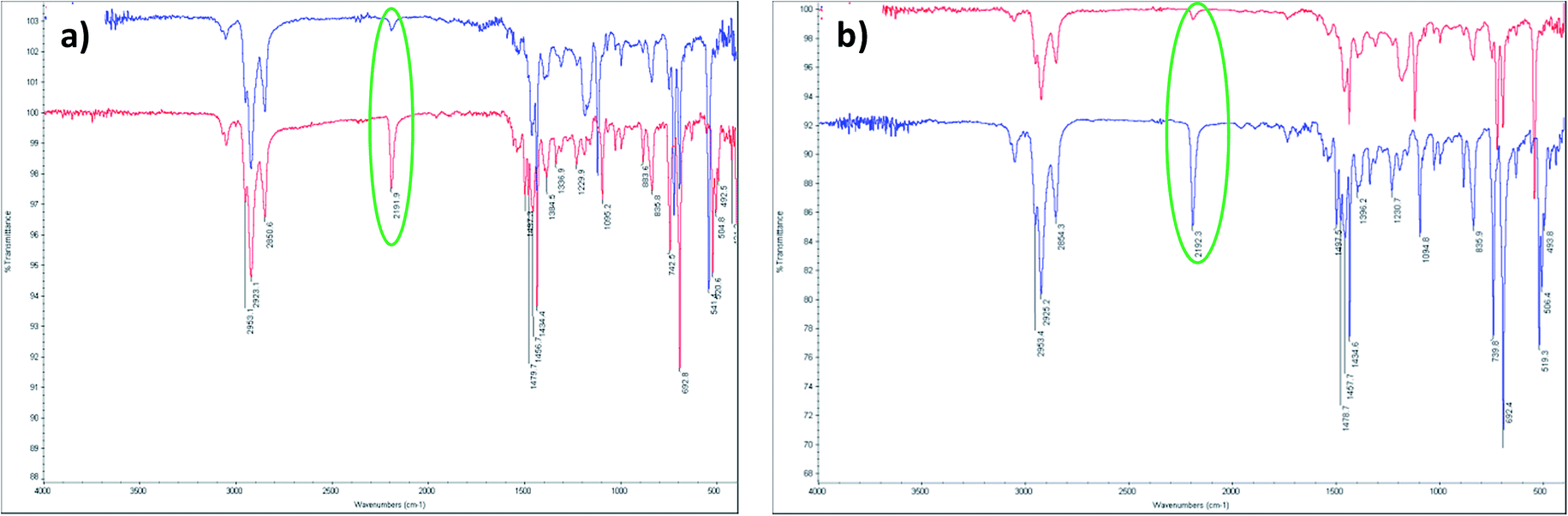 | ||
| Fig. 2 Fourier transform infrared (FT-IR) spectra of polymers (a) D–A and (b) A–A before (under line) and after (above line) UV treatment. | ||
3.3. Electrochemical properties
To determine the energy levels of the synthesized polymers, cyclic voltammetry (CV) is used. The results are shown in Fig. 3 and Table 1. For calibration, the redox potential of ferrocene/ferrocenium (Fc/Fc+) is measured and it is located at 0.22 V to the Ag/AgNO3 (0.1 M) electrode. It is assumed that the redox potential of Fc/Fc+ has an absolute energy level of −4.8 eV to vacuum.15 Then the energy level of the highest occupied molecular orbital (HOMO) is calculated according to the following equation: EHOMO = −(4.58 + Eox onset) (eV) and the lowest unoccupied molecular orbital (LUMO) is calculated from EHOMO and the optical band gap according to the following equation: ELUMO = (EHOMO + Eoptg) (eV). The first oxidation potential of D–A appeared at Eox = +1.38 V (versus Ag/Ag+) while in the case of A–A, it appeared at Eox = +0.75 V (versus Ag/AgNO3 (0.1 M)), corresponding to HOMO energy levels for D–A and A–A of −5.96 and −5.33 eV, respectively. Meanwhile, the LUMO energy levels of D–A and A–A were calculated to be −3.71 and −3.43 eV, respectively.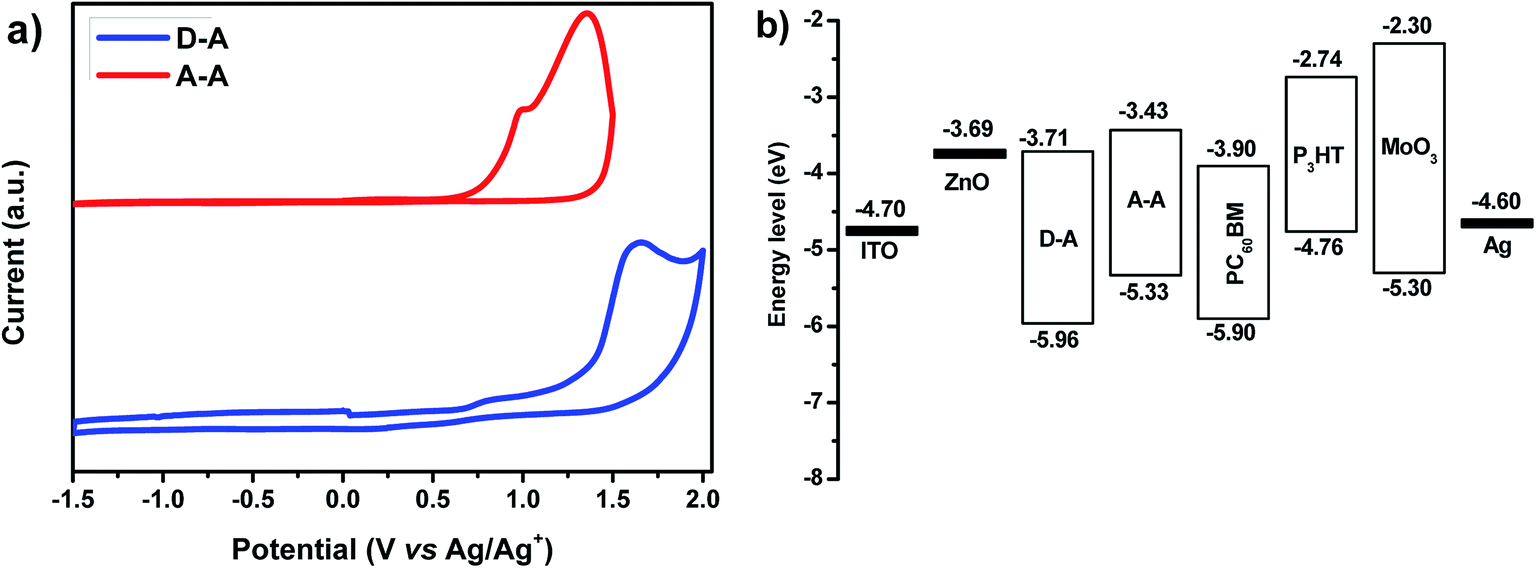 | ||
| Fig. 3 (a) Cyclic voltammograms and (b) energy levels of the device components. | ||
3.4. The effect of the buffer layer on the properties of the ZnO surface
To characterize the ZnO surface before and after using a buffer layer with UV treatment, AFM images were measured and are shown in Fig. 4. The roughness of the ZnO surface was not greatly affected by coating with the A–A polymer buffer layer; the root mean square (RMS) roughness values of neat ZnO, ZnO with an A–A buffer layer, and ZnO with an A–A buffer layer with UV exposure are 0.927, 1.080, and 0.996 nm, respectively. However, in the case of the D–A-based buffer layer, the roughness was significantly increased with values of 1.551 and 1.990 nm, respectively, for without and with UV irradiation after D–A polymer introduction on top of the ZnO surfaces. The surface of the A–A polymer introduced buffer layer showed a fabric network after UV exposure which may be formed from the A–A crosslinked polymers, while in the case of the D–A polymer, big grains were formed, which led to significantly increased roughness. | ||
| Fig. 4 (a, c, e, g, i) for height images and (b, d, f, h, j) for phase AFM images of pristine ZnO surface and polymer coated ZnO surfaces without and with UV treatment. | ||
The hydrophilicity of the ZnO surface was compared before and after spin-coating with a buffer layer via contact angle measurement using water drops, as shown in Fig. 5. The contact angle was changed from 55.27° to 86.81° (with the A–A polymer) and to 75.24° (with the D–A polymer) when a buffer layer was applied on top of the ZnO. After UV exposure, the A–A crosslinked polymer has a contact angle value of 82.65°, which is not much different from before UV irradiation, while in the case of the D–A crosslinked polymer, the contact angle clearly decreased to 66.69°. The maintained hydrophobic and smoother surface of the A–A polymer was caused by good surface coverage of the crosslinked buffer layer, but the reduced hydrophobicity and increased surface roughness of the D–A polymer means the surface of the ZnO was not fully covered with the buffer layer. This poorly distributed buffer layer of polymer D–A may reduce the FF of the devices.
 | ||
| Fig. 5 Images of the water contact angle of neat ZnO and treated ZnO. | ||
It is well known that the presence of buffer layers in PSCs affects the work function (WF) of the electrodes.16 To investigate this effect, the WFs of the neat ZnO, polymer coated ZnO, and polymer coated ZnO after UV treatment were measured using ultraviolet photoelectron spectroscopy (UPS), as shown in Fig. 6. The WF value of neat ZnO was calculated to be 3.69 eV, while the WF values of ZnO coated with polymers D–A and A–A were increased to 3.75 and 3.70 eV, respectively, and further increased to 3.95 and 3.85 eV, respectively, after 5 min of UV exposure. This result means that the WF of the cathode has been down-shifted after coating with polymers and UV exposure.
 | ||
| Fig. 6 Work functions (a) and Fermi-edge region (b) of neat ZnO and polymer coated ZnO without and with UV treatment determined by UPS studies with a He (hν = 21.2 eV) source. | ||
3.5. Photovoltaic characteristics
To study the effect of crosslinked polymers on the photovoltaic performance of inverted OSCs, two kinds of inverted PSCs were fabricated with the configuration of ITO/ZnO/active layer/MoOx(10 nm)/Ag(100 nm) and ITO/ZnO/crosslinked polymer/active layer/MoOx(10 nm)/Ag(100 nm). The active layer materials used were P3HT as a donor and PC60BM as an acceptor with a weight ratio of 1:0.8 in o-DCB. CB solvent was used to make the solution of crosslinkable polymers. The optimal solution concentrations to spin-cast the buffer layer were investigated and found to be 2 mg mL−1 for polymer D–A and 4 mg mL−1 for polymer A–A. Fig. 7a and b show the J–V curves of PSCs under the conditions of AM 1.5 at 100 mW cm−2, and the open circuit voltage (VOC), short-circuit current density (JSC), FF, and PCE values are summarized in Table 2.
 | ||
| Fig. 7 (a, b) J–V curves and (c, d) EQE spectra of BHJ solar cells with the device structure ITO/ZnO/active layer/MoOx(10 nm)/Ag(100 nm) and ITO/ZnO/polymer/active layer/MoOx(10 nm)/Ag(100 nm); (a, c) polymer D–A and (b, d) polymer A–A. | ||
| Material & conc. | UV irrad. time [min] | VOC [V] | JSC [mA cm−2] | JEQESCa [mA cm−2] | FF [%] | PCEb [%] |
|---|---|---|---|---|---|---|
| a Calculated by integrating the EQE spectrum with the AM1.5G spectrum.b The average PCE was obtained from over 8 devices. | ||||||
| Reference | x | 0.61 ± 0.002 | 8.94 ± 0.559 | 8.43 | 50 ± 2.2 | 2.74 ± 0.181 |
| A–A 4 mg mL−1 | 0 | 0.61 ± 0.000 | 8.15 ± 0.160 | 8.86 | 56 ± 2.0 | 2.78 ± 0.070 |
| 5 | 0.61 ± 0.000 | 8.40 ± 0.130 | 8.99 | 60 ± 3.0 | 3.10 ± 0.160 | |
| D–A 2 mg mL−1 | 0 | 0.62 ± 0.003 | 8.90 ± 0.510 | 8.59 | 53 ± 2.7 | 2.91 ± 0.278 |
| 5 | 0.62 ± 0.001 | 8.81 ± 0.530 | 8.42 | 41 ± 1.5 | 2.25 ± 0.016 | |
The PSC with a bare ZnO layer showed an average PCE value of 2.74% with a VOC of 0.61 V, a JSC of 8.94 mA cm−2, and a FF of 50%. With the D–A polymer interlayer, the PCE of the device was improved to 2.91% with a VOC of 0.62 V, a JSC of 8.90 mA cm−2, and a FF of 53%, even without UV exposure. On the other hand, in the case of the non-UV treated A–A polymer interlayer device, the PCE was only slightly increased with a value of 2.78%, a VOC of 0.61 V, a JSC of 8.15 mA cm−2, and a FF of 56%. Overall, even without crosslinking, the average PCE were increased in both the A–A and D–A polymer devices in accordance with the improved FF. To investigate the effect of the crosslinked polymer buffer on the PSC performance, the A–A and D–A polymer layers were exposed to a UV lamp for 5 min after spin-coating. In the case of the D–A polymer based device, the average PCE dropped significantly to 2.25% after 5 min UV irradiation of the buffer layer, mainly caused by the sharp reduction of FF from 53% to 41%. In contrast with the trend for the D–A polymer, UV treatment of the A–A polymer increased the PCE of the PSC devices. After 5 min of UV treatment, the PSC performance of the A–A polymer based device showed a PCE value of 3.10% with an increased JSC and FF.
In the literature, there are many reports on the effect of the work function of two electrodes on the VOC value,17–19 and the shifting of energy levels of the electrodes was used to explain the change in VOC values of the PSCs. However, in the case of ZnO with a triple bond coating, although the work function of the ZnO buffer layer was changed after coating with the triple bond polymer (without and with UV treatment), the VOC was not significantly affected. This phenomenon recently was reported and explained by the lower conductivity of the metal oxide (ZnO) layer than those of metal and transparent electrodes.20
4. Conclusions
Three novel triple bond containing polymers, named A–A, D–A and D–D, were designed, and two of them, polymers A–A and D–A, were investigated for their photocrosslinkability, as well as their application as buffer layers in OPVs. The occurrence of crosslinking was proven using UV-vis spectroscopy and IR techniques. The PSC device with an A–A polymer buffer layer exhibited the best average PCE of 3.10% after 5 min of UV exposure, caused by the improved FF compared to the device with a bare ZnO layer. Our research introduced potential candidates for new photocrosslinkable materials which have solvent resistance and a hydrophobic nature, and can be used in solution processed multilayer organic electronic devices.Acknowledgements
This research was supported by the New & Renewable Energy Core Technology Program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), and granted financial resources from the Ministry of Trade, Industry & Energy (No. 20133030011330) and the Technology Development Program to Solve Climate Changes of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2015M1A2A2056214), Republic of Korea.References
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nature Energy, 2016, 1, 15027 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- T. R. Hebner, C. C. Wu, D. Marcy, M. H. Lu and J. C. Sturm, Appl. Phys. Lett., 1998, 72, 519–521 CrossRef CAS.
- C. A. Zuniga, S. Barlow and S. R. Marder, Chem. Mater., 2011, 23, 658–681 CrossRef CAS.
- C. Z. Li, H. L. Yip and A. K. Y. Jen, J. Mater. Chem., 2012, 22, 4161–4177 RSC.
- N. S. Kang, B. K. Ju, T. W. Lee, D. H. Choi, J. M. Hong and J. W. Yu, Sol. Energy Mater. Sol. Cells, 2011, 95, 2831–2836 CrossRef CAS.
- Y. Udum, P. Denk, G. Adam, D. H. Apaydin, A. Nevosad, C. Teichert, M. S. White, N. S. Sariciftci and M. C. Scharber, Org. Electron., 2014, 15, 997–1001 CrossRef CAS PubMed.
- M. Kato and Y. Yoneshige, J. Polym. Sci., Polym. Lett. Ed., 1979, 17, 79–83 CrossRef CAS.
- A. S. Hay, D. A. Bolon and K. R. Leimer, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 1022–1023 CrossRef CAS.
- M. C. Hung, J. L. Liao, S. A. Chen, S. H. Chen and A. C. Su, J. Am. Chem. Soc., 2005, 127, 14576–14577 CrossRef CAS PubMed.
- X. Huang, Y. Xu, L. Zheng, J. Meng and Y. Cheng, Polymer, 2009, 50, 5996–6000 CrossRef CAS.
- M. He, T. M. Leslie and J. A. Sinicropi, Chem. Mater., 2002, 14, 4662–4668 CrossRef CAS.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Baessler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- C. E. Song, K. Y. Ryu, S. J. Hong, C. Bathula, S. K. Lee, W. S. Shin, J. C. Lee, S. K. Choi, J. H. Kim and S. J. Moon, ChemSusChem, 2013, 6, 1445–1454 CrossRef CAS PubMed.
- C. Waldauf, M. Morana, P. Denk, P. Schilinsky, K. Coakley, S. A. Choulis and C. J. Brabec, Appl. Phys. Lett., 2006, 89, 233517 CrossRef.
- Z. Tan, W. Zhang, Z. Zhang, D. Qian, Y. Huang, J. Hou and Y. Li, Adv. Mater., 2012, 24, 1476–1481 CrossRef CAS PubMed.
- S. K. Hau, H. L. Yip, H. Ma and A. K. Y. Jen, Appl. Phys. Lett., 2008, 93, 233304 CrossRef.
- S. Nho, G. Baek, S. Park, B. R. Lee, M. J. Cha, D. C. Lim, J. H. Seo, S. H. Oh, M. H. Song and S. Cho, Energy Environ. Sci., 2016, 9, 240–246 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |