
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High capacitive amorphous barium nickel phosphate nanofibers for electrochemical energy storage†
Teng
Wang
a,
Qingli
Hao
b,
Jinzhang
Liu
c,
Jiachang
Zhao
d,
John
Bell
a and
Hongxia
Wang
*a
aSchool of Chemistry, Physics and Mechanical Engineering, Science and Engineering Faculty, Queensland University of Technology, Brisbane, QLD 4001, Australia. E-mail: hx.wang@qut.edu.au
bKey Laboratory for Soft Chemistry and Functional Materials, School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing, China
cSchool of Materials Science and Engineering, Beihang University, Beijing, China
dCollege of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, Shanghai, China
First published on 4th May 2016
Abstract
Ultrafine amorphous BaxNi3−x(PO4)2 (0 < x < 3) nanofibers are synthesized for the first time through a facile cation exchange reaction method at room temperature. Both the phase transformation and growing process of the nanofibers are systematically investigated. A dramatic morphology transformation from Ba3(PO4)2 flakes to BaxNi3−x(PO4)2 nanofibers with the addition of Ni2+ ions is observed. The as-prepared nanofiber material shows a diameter less than 10 nanometers and length of several micrometers. The material possesses a BET surface area of 64.8 m2 g−1. When it is used as a supercapacitor electrode material, specific capacitances as high as 1058 F g−1 at 0.5 A g−1 and 713 F g−1 at 5 A g−1 are achieved, indicating the promising energy storage property of this material.
Introduction
Materials based on metal phosphates have attracted attention in the scientific community due to their promising performance when used in varying applications including sensors, gas adsorption/desorption, supercapacitors, optoelectronics, lithium-ion batteries etc.1–8 Attfield et al. firstly synthesized a series of metal phosphates with a molecular formula of M11(HPO3)8(OH)6 (M = Zn, Co, or Ni) through soft hydrothermal treatments.9 Inspired by this work, various phosphates containing different cations or cation combinations like nickel, cobalt, ammonium and sodium, have been synthesized by hydrothermal method.10–15 Most metal phosphates have a layered structure. This unique structure characteristic is desired for applications such as energy storage devices which require fast transport of ions.Supercapacitors, also called electrochemical capacitors, are one of the most efficient energy storage devices that have been used in practice due to their advantages of high power density and ultra-long cycle life.16–20 However, current commercial supercapacitors suffer from a limited energy density (less than 10 W h kg−1), which prevents their broader application in practice.21 One of the methods to improve the energy density of supercapacitors is by employing nanostructured pseudocapacitive electrode materials which possess larger specific surface areas, more active sites and shorter transport/diffusion path for electrolytic ions and electrons compared to bulk materials.22
Besides metal oxides which are widely studied as pseudocapacitive materials,23–27 limited research has shown that one-dimensional (1D) phosphate materials can also be effective supercapacitive materials. Pang et al. reported the supercapacitive performance of phosphite materials based on (M11(HPO3)8(OH)6) (M = Co, Ni)28,29 and (Ni20[(OH)12(H2O)6]·[(HPO4)8(PO4)4]·12H2O)30 which were made by hydrothermal method. Benefiting from the high pressure and heating of hydrothermal reaction condition, the material showed good crystallinity and exhibited higher energy density compared to conventional carbon based materials.
However, more recent studies have shown that amorphous state of nanomaterials may be more beneficial in supercapacitor devices compared to their crystallinity counterpart owing to the higher structural disorder, larger density of active site of the former, leading to higher energy density. Yang et al. reported amorphous Ni(OH)2 nanospheres whose capacitive performance was commensurate with the crystalline materials.31 Moreover, Cao et al. reported the supercapacitive performance of ultrathin amorphous Co3(PO4)2 nanowires which exhibited higher capacitance than the crystalline counterpart material.32
Herein, we report the synthesis of a new phosphate material based on amorphous BaxNi3−x(PO4)2 nanofibers with diameter less than 10 nanometres through a facile cation exchange reaction method in aqueous solution at room temperature. By monitoring the phase transformation and growth process of BaxNi3−x(PO4)2 material by SEM and XRD, we have observed a clear morphology evolution from Ba3(PO4)2 microplates to microflowers and finally to BaxNi3−x(PO4)2 nanofibers with the increased concentration of Ni(NO3)2 in the precursor solution. A growth mechanism which involves self-assembly of Ba3(PO4)2 microplates that gradually transformed into BaxNi3−x(PO4)2 nanofibers is proposed. The study of argon adsorption–desorption isotherm shows the as-prepared nanofibers has a large BET surface area of 64.8 m2 g−1, which provides sufficient active sites for the faradaic charge transfer process when used as electrode materials of SCs. The maximum specific capacitance of 1058 F g−1 (at 0.5 A g−1) and 713 F g−1 (at 5 A g−1) were obtained, which is much higher than the corresponding crystalline material made by annealing the material at 800 °C. The remarkable capacitance and good electrochemical properties of BaxNi3−x(PO4)2 material are attributed to the unique amorphous nanostructure and the efficient faradaic charge transfer process involving the reversible redox reaction of nickel cations.
Experimental
Materials
All the chemicals of analytical grade were purchased from Alfa Aesar and used as received without any further purification, unless otherwise stated. Deionized water with a resistivity of 18.2 MΩ cm was used in all reactions. Nickel foams were provided by Sigma-Aldrich.Preparation of BaxNi3−x(PO4)2 nanofibers
BaxNi3−x(PO4)2 nanofibers were prepared through a facile solution method based on cation exchange reaction. In a standard synthesis procedure, 20 mM Na3PO4·12H2O aqueous solution was dropwise added into Ba(NO3)2 aqueous solution (30 mM) under continuous magnetic stirring. A white precipitate was formed gradually during this process. After 2 min reaction, 30 mM Ni(NO3)2·6H2O aqueous solution was further dropwise added into above reaction solution. After 48 h reaction under continuous vigorous stirring, all white precipitates were found to be transformed into light green colour, which was then collected through centrifuge. The material was rinsed at least three times with large amount of deionized water before being dried at 80 °C in an electrical oven.Material characterizations
The morphology and elemental distributions of the products were characterized by field emission scanning electron microscope (FESEM, JSM-7001F, JEOL) employing an accelerating voltage of 5.00 kV with an energy dispersive spectrometer (EDS). Transmission electron microscope was used to measure the crystallinity and morphology of as-prepared materials (TEM, JEOL 2100). X-ray diffraction analysis (XRD, PANaytical MPD Cu Powder XRD) using Cu Kα radiation at 40 keV and 40 mA was employed to determine the crystalline structure and composition of the as-prepared products. X-ray photoelectron spectroscopy (XPS) was used to obtain the elemental composition and valence state, which was performed by a photoelectron spectrometer using a non-monochromatic Mg Kα (1253.6 eV) X-ray source (DAR 400, Omicron Nanotechnology) with incident angle at 65° to the sample surface. The signal was collected by a 125 mm hemispherical electron energy analyser (Sphera II, 7 channels detector, Omicron Nano-technology). Micromeritics Tristar II 3020 Surface Area Analyser was used to test the BET surface area of the sample.Electrochemical measurements
The electrochemical properties of BaxNi3−x(PO4)2 nanofibers were comprehensively investigated using a three-electrode configuration cell with as-synthesized material as working electrode, Pt wire as counter electrode and calomel electrode as reference in 2.0 M KOH aqueous electrolyte. The working electrodes were fabricated by following a similar procedure reported by other groups previously.33 Firstly, a mixture of BaxNi3−x(PO4)2 nanofibers, carbon black, and poly(vinylidene fluoride) (PVDF) with mass ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 was dissolved in N-methylpyrrolidinone (NMP) to make a slurry under continuous stirring for 24 h, which was then pasted on a nickel foam (1 × 1 cm2). After drying in a vacuum oven at 110 °C for 10 h, the coated nickel foam was pressed into a thin film under 10 MPa pressure. Each working electrode contained 1–2 mg active material. All the electrochemical properties were performed with an electrochemical workstation (SP-150, BioLogic Science Instruments) at room temperature (22 °C). The electrochemical impedance spectral (EIS) measurements were carried out in the frequency range of 100 kHz to 10 mHz with an AC amplitude of 5 mV.
:10:10 was dissolved in N-methylpyrrolidinone (NMP) to make a slurry under continuous stirring for 24 h, which was then pasted on a nickel foam (1 × 1 cm2). After drying in a vacuum oven at 110 °C for 10 h, the coated nickel foam was pressed into a thin film under 10 MPa pressure. Each working electrode contained 1–2 mg active material. All the electrochemical properties were performed with an electrochemical workstation (SP-150, BioLogic Science Instruments) at room temperature (22 °C). The electrochemical impedance spectral (EIS) measurements were carried out in the frequency range of 100 kHz to 10 mHz with an AC amplitude of 5 mV.
The specific capacitance (CT) of the electrode is calculated based on the measured galvanostatic charging/discharging (GCD) curves, according to eqn (1):34–36
 | (1) |
Results and discussion
As stated in experimental section, a white precipitate was firstly observed by addition of Na3PO4 to Ba(NO3)2, which then transformed to light green color by further addition of Ni(NO3)2·6H2O. In order to understand this process, the XRD and SEM of both the white precipitate and the final product of light green precipitate were recorded. The results show that the white precipitate is barium phosphate (Ba3(PO4)2, Fig. S1(a)†), which adopts rhombohedral crystal phase (PDF#80-1615) and shows good crystallinity. SEM image shows the barium phosphate has a regular shape of hexagon plate with size of several micro-meters (Fig. S1(b)†). The nature of Ba3(PO4)2 white precipitate is also confirmed by the selected area electron diffraction (SAED) pattern (Fig. S1(c)†) and high resolution transmission electron microscope (HRTEM, Fig. S1(d)†).The SEM images of the light green precipitate shown in Fig. 1(a and b) confirm that the final products consist of nanofibers which are micrometer in length and nanometer in width. The TEM images (shown in Fig. 1(c and d)) further reveal that the nanofibers are bundled together and each fiber is less than ten nanometers in diameter. The as-prepared nanofibers show an amorphous nature because no lattice fringe could be detected by the HRTEM image as shown in Fig. 2. The corresponding SAED pattern further confirms their amorphous structure (inset in Fig. 2). In order to confirm the elemental composition of as-prepared nanofibers, EDS was carried out and the result is shown in Fig. S2 (ESI†). Elements including Ba, Ni, P, and O are clearly detected in the EDS profile with the ratio of Ba/Ni = 4:7, confirming the chemical composition of the obtained material. Above result confirms the partial replacement of Ba2+ in Ba3(PO4)2 by Ni2+ during the transformation from white precipitate to light green product.
 | ||
| Fig. 1 (a and b) SEM images of BaxNi3−x(PO4)2 nanofibers; (c and d) regular and enlarged TEM images of BaxNi3−x(PO4)2 nanofibers. | ||
 | ||
| Fig. 2 HRTEM image and corresponded SAED pattern of BaxNi3−x(PO4)2 nanofibers. | ||
The XRD patterns of as-prepared BaxNi3−x(PO4)2 are shown in Fig. 3. Apparently, only a broad and weak diffraction pattern is observed with the fresh product (named as BaNiP), which is in agreement with the HRTEM result shown in Fig. 2, confirming the amorphous nature of the as-synthesized material. This is probably related with the low synthesis temperature process used. To confirm this, a high temperature calcination was employed to investigate the crystallization process of as-synthesized materials. The XRD of the material annealed at 350 °C for 2 h in air (referred as BaNiP-350, green line) shows that the broad diffraction peak in BaNiP becomes narrower. Meanwhile, several additional broad peaks are observed in the XRD pattern of BaNiP-350, which suggests the crystallinity of the material is improved with the annealing at 350 °C but it is still poor. By further increasing the annealing temperature to 800 °C, the XRD peaks of the obtained product (named as BaNiP-800) are strong and sharp. All the peaks of the XRD pattern can be well indexed to the crystal structure of rhombohedral BaNi2(PO4)2 (ICSD-01-089-6720). The absence of other peaks detected in the XRD pattern of BaNi-800 demonstrates the high purity of the material.
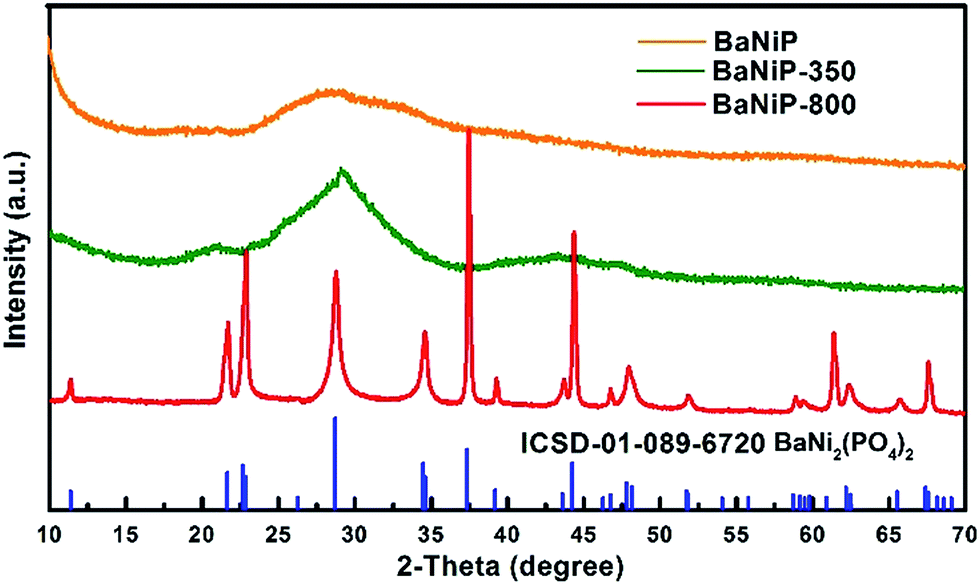 | ||
| Fig. 3 XRD patterns of fresh BaxNi3−x(PO4)2 nanofibers and annealed products at different temperature (350 °C and 800 °C). | ||
Furthermore, XPS was employed to analyse the surface composition and oxidation state of BaxNi3−x(PO4)2 nanofibres, in which C 1s (284.8 eV) was used to calibrate the binding energies. As shown in Fig. 4(a), the full range XPS spectrum clearly shows the characteristic peaks of Ni, Ba, P, O and C elements. It is worth noting that the C element comes from the residual carbon-based contaminants instead of the as-prepared BaxNi3−x(PO4)2 nanofibres. The high resolution XPS (HRXPS) spectrum of Ni 2p (Fig. 4(b)) shows the binding energy peaks at 872.7 eV and 855.0 eV, which are corresponding to Ni 2p1/2 and Ni 2p3/2, respectively, indicating that nickel cations are Ni2+ binding state.37 It is worth to note that the intensive shoulder peaks (860.1 and 879.1 eV, marked as Sat 1 and Sat 2) along with the main peaks of Ni 2p3/2 and Ni 2p1/2 are due to two shakeup-type peaks of nickel.38Fig. 4(c) presents the HRXPS spectrum of Ba 3d1/2 and Ba 3d5/2 with the characteristic peaks detected at 795.0 eV and 779.6 eV, respectively. The peak positions and binding energy gap between the two signals clearly demonstrate the existence of Ba2+.39–41 Moreover, the HRXPS spectra in Fig. 4(d) and (e) show the state P 2p and O 1s with the binding energy at 132.8 and 530.7 eV, respectively, further confirming the formation of BaxNi3−x(PO4)2 nanomaterials.42
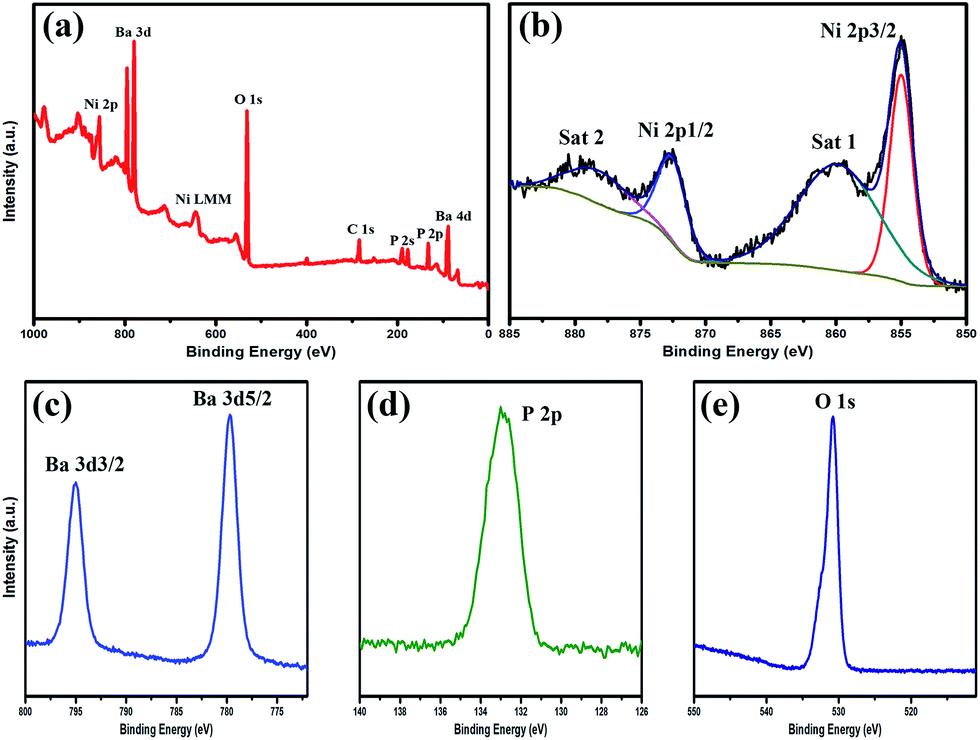 | ||
| Fig. 4 (a) Full range XPS spectrum of BaxNi3−x(PO4)2 nanofibers; (b–e) HRXPS spectra of Ba, Ni, P, and O elements. | ||
During the process of fabrication of BaxNi3−x(PO4)2 nanofiber, we noticed that the content of precursor Ni(NO3)2 plays a key role in the formation of nanofibers. Fig. 5 and S3† show the SEM images and XRD patterns of BaxNi3−x(PO4)2 material synthesized with different concentration of Ni(NO3)2. When the content of Ni(NO3)2 is only 5 mM, the product consists of material with microflower structure (Fig. 5(a)), which is believed due to the self-assembly of partially reacted Ba3(PO4)2 flakes. By increasing the concentration of Ni(NO3)2 solution to 10 mM, the mircroflowers are disassembled and lots of flakes are found in the material (Fig. 5(b)). Further increase the concentration of Ni(NO3)2 to 20 mM in the precursor solution, nanofibers start to form among the main morphology of flakes which are interweaved together as indicated in Fig. 5(c). The flakes are completely transformed into nanofibers when 30 mM Ni(NO3)2 is used in the reaction solution (Fig. 5(d)). Beyond this, the morphology of the material does not change. XRD results (Fig. S3†) confirm strong diffraction peaks of Ba3(PO4)2 flakes when 10 mM Ni(NO3)2 is used in the reaction. A complete transformation from Ba3(PO4)2 into BaxNi3−x(PO4)2 nanofibers occurs when the concentration of Ni(NO3)2 solution is over 30 mM along with the disappearance of the characteristic XRD peaks of Ba3(PO4)2.
 | ||
| Fig. 5 SEM images of BaxNi3−x(PO4)2 material synthesized from different concentration of Ni(NO3)2 of 5 mM (a), 10 mM (b), 20 mM (c), 30 mM (d). | ||
In order to further understand the formation mechanism of the nanofibers, the SEM images of BaxNi3−x(PO4)2 with different reaction time were recorded and are shown in Fig. 6. It can be seen that the smooth surface of pristine Ba(PO4)2 material becomes rough within only 5 min reaction (Fig. 6(a)). Further elongation of the reaction duration, Ba3(PO4)2 microplates turn into fluffy and some of them merge together to form thin films gradually (Fig. 6(b)). Nanofibers start to form on the amorphous films after 2 h reaction (Fig. 6(c) and inset). Further extending the reaction to 48 h leads to the full transformation of all the material into nanofibers (Fig. 6(d)).
 | ||
| Fig. 6 SEM images of BaxNi3−x(PO4)2 materials with different reaction time of 5 min (a), 30 min (b), 2 h (c) and 48 h (d). | ||
Based on the above results, the formation mechanism of the nanofibers is proposed as shown in Scheme 1. It is known that Ba3(PO4)2 adopts a layer-structure with a characteristic of rod sequence of polyhedral in the form of PO4–Ba(2)O10–Ba(1)O12–Ba(2)O10–PO4 along the c axis.43–45 The weak bonding strength between Ba2+ and oxygen makes Ba2+ easy to be substituted by other divalent cations. This explains the partial substitution of Ba2+ by Ni2+ in the cations exchange process. In this process, the added Ni2+ firstly intercalates into the interlayer of the microplated structure of Ba3(PO4)2. The subsequent cation exchange reaction results in the formation of a rough surface of the original Ba3(PO4)2 flakes in the first few minutes of reaction. As more Ni2+ ions intercalate into the host, the layered structure is expanded and finally set apart into thin layered films (Fig. 6(b)). Sufficient Ni2+ ions are critical in this process to ensure fully penetration and separation of all the pristine Ba3(PO4)2 layers. It is expected that the replacement of Ba2+ should occur along the c axis direction by considering the molecule structure of Ba3(PO4)2, which is responsible for the formation of nanofiber structure (Fig. 6(c)). The long intensively magnetic stirring further facilitates split of the weakly bonded films into nanofibers.
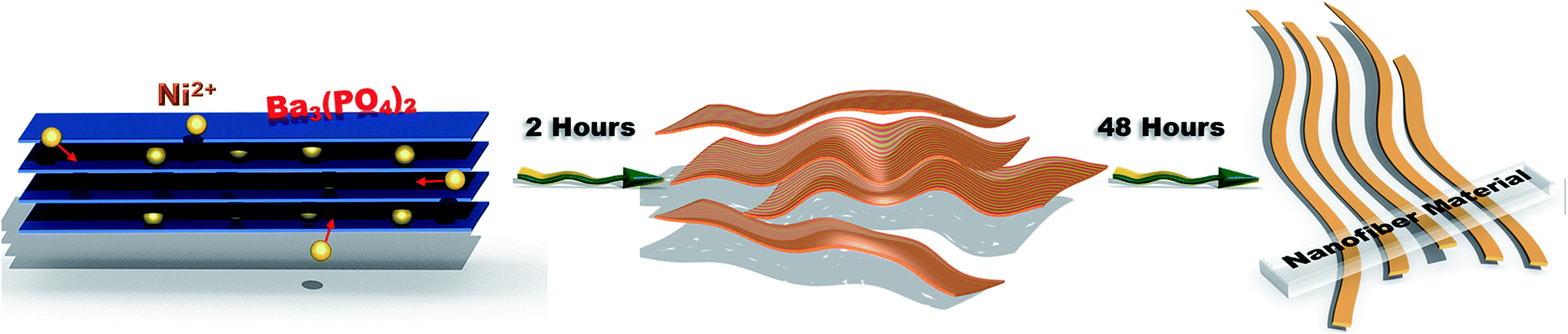 | ||
| Scheme 1 Schematic illustration of the formation of BaxNi3−x(PO4)2 nanofiber. | ||
The energy storage properties of as-synthesized BaxNi3−x(PO4)2 nanofibers were investigated in a typical three-electrode electrochemical system. The cyclic voltammetry (CV) and GCD plots of BaxNi3−x(PO4)2 nanofiber electrode are shown in Fig. 7(a and b), respectively. It can be seen from Fig. 7(a) that a distinct pair of oxidation and reduction peaks can be clearly observed in the CV plots with different scan rates, demonstrating a strong pseudocapacitive behaviour.46 The pseudocapacitance should be originated from the redox reaction of Ni2+/Ni3+ during the faradaic charge transfer process other than Ba2+ considering the sole valence state of the cations of barium.47,48 Moreover, the high current response in the CV curves indicates that BaxNi3−x(PO4)2 nanofibers possess considerable charge storage capability.
 | ||
| Fig. 7 (a and b) The cyclic voltammetry and galvanostatic charging/discharging curves of BaxNi3−x(PO4)2 nanofiber electrode in 2 M KOH electrolyte, respectively; (c) calculated specific capacitance of BaxNi3−x(PO4)2; (d) Nyquist plot of the impedance of BaxNi3−x(PO4)2 electrode from 0.01 Hz to 100 kHz. | ||
The GCD plots under different charging current densities (Fig. 7(b)) further confirm the pseudo-capacitive behaviours of the electrode material with two distinct plateaus during the charging/discharging process. The specific capacitance of BaxNi3−x(PO4)2 nanofiber electrode under various discharging current densities was calculated according to the GCD curves. As shown in Fig. 7(c), the specific capacitance of the electrode is 1058, 968, 870, 713, 575 and 400 F g−1 at the discharging current density of 0.5, 1, 2, 5, 10 and 20 A g−1, respectively. Clearly, the electrode retains over 50% of the initial capacitance even when the discharging current density is increased by 20 times from 0.5 A g−1 to 10 A g−1, indicating a good rate capability. This is ascribed to the high conductivity of the material as confirmed by electrochemical impedance spectrum (EIS) of the electrode in Fig. 7(d). The EIS data was fitted using an equivalent circuit by Zview software. As shown in the inset of Fig. 7(d), the equivalent circuit consists of a series resistance Rs, a charge-transfer resistance Rct, and constant phase elements CPE and CPE1 corresponding to a pseudocapacitance of the electrode and double-layer capacitance at the electrode material/electrolyte interface, respectively.49 The EIS fitting results show that the synthesized BaxNi3−x(PO4)2 nanofiber electrode has a low series resistance (Rs = 0.80 Ω) and charge transfer resistance (Rct = 0.56 Ω), which is believed to be one of the reasons for the excellent supercapacitive performance of the material.
The cycling performance of the as-prepared electrode is also studied. As shown in Fig. 8, around 80% of the initial capacitance of the electrode is retained after 500 cycles at a discharging current of 3 A g−1, indicating a relatively good cycling stability. However compared to other reported crystalline phosphates materials,27–29 further improvement of the cycling stability of the material is needed. In order to understand the reason for the unsatisfactory stability of the material, the SEM image of the working material after 500 cycles was recorded (Fig. S4†). It is found that the nanofiber structure of the synthesized BaxNi3−x(PO4)2 is destroyed after the stability test. This suggests a large volume change occurring within the material during the charging/discharging process, which results in the broken of nanofiber structure into pieces. Previous work by other researchers have shown that incorporation of more conductive and stable materials such as carbon materials or metal oxides can normally improve the stability and electrochemical performance of non-carbon materials.50,51 This will be the subject of our future research.
 | ||
| Fig. 8 Cycling stability test of BaxNi3−x(PO4)2 material synthesized from 30 mM Ni(NO3)2. | ||
The remarkable supercapacitive performance of the synthesized BaxNi3−x(PO4)2 nanofibers is attributed to the unique amorphous nanostructure and large surface area. The argon adsorption–desorption isotherm plot reveals that BaxNi3−x(PO4)2 nanofibers have a high BET surface area of 64.8 m2 g−1 (Fig. 9). As shown in Fig. S5,† the amorphous sample owns a 3 times higher current peak than the crystalline material (800 °C annealed sample) in the cyclic voltammetry measurement, confirming the superior energy storage property of the amorphous structure. Similar phenomenon has been reported by Li et al. who found superior supercapacitive performance with a series of amorphous Ni–Co–Fe hydroxides.52 It is ascribed to the unique nature of amorphous materials which is disorder in long-range but is structurally order in short-range. Moreover amorphous material possesses much more unsaturated ligand atoms and efficient electrochemically active sites compared to crystalline counterpart.52 These features favour the charge transport process in the material. This is confirmed by the low interface charge transfer resistance and good rate ability of the BaxNi3−x(PO4)2 material shown above. Moreover, the relatively large surface area provides sufficient pathway for the transport of charged ions, which further boosts the supercapacitive performance of the material.
 | ||
| Fig. 9 Argon adsorption–desorption isotherm linear plot of BaxNi3−x(PO4)2 material. | ||
Conclusions
In summary, nanofiber-structured amorphous BaxNi3−x(PO4)2 material has been synthesized for the first time through a facile cation-exchange reaction method at room temperature. The as-prepared materials exhibited typical pseudocapacitive behaviour with high electrical storage capacity. The specific capacitance of 1058 F g−1 at a discharging current density of 0.5 A g−1 was achieved. The good electrochemical properties of BaxNi3−x(PO4)2 materials are attributed to the ultrafine amorphous nanofiber structure and high surface area of 64.8 m2 g−1. Just as other pseudocapacitive materials, the cycling stability of individual BaxNi3−x(PO4)2 material also needs to be improved for further application in supercapacitors. The investigation of the formation mechanism shows that the concentration of precursor Ni(NO3)2 solution plays a critical role in the formation of the nanofiber structure. The method of synthesizing nanostructured phosphate materials using cation exchange reaction paves a facile way for synthesis of materials with controlled morphology and good electrochemical properties to meet the high-energy density requirement of practical applications in energy storage systems, such as high-performance supercapacitors.Acknowledgements
H. W. acknowledges financial support from the Australian Research Council (ARC) Future Fellowship (FT120100674) for this work. T. W. thanks Queensland University of Technology (QUT) for Postgraduate Research Award scholarships.Notes and references
- C. Wu, X. Lu, L. Peng, K. Xu, X. Peng, J. Huang, G. Yu and Y. Xie, Nat. Commun., 2013, 4, 2431 Search PubMed.
- J.-H. Yang, J. Tan and D. Ma, J. Power Sources, 2014, 260, 169–173 CrossRef CAS.
- C. Sun, S. Rajasekhara, J. B. Goodenough and F. Zhou, J. Am. Chem. Soc., 2011, 133, 2132–2135 CrossRef CAS PubMed.
- C. Chen, W. Chen, J. Lu, D. Chu, Z. Huo, Q. Peng and Y. Li, Angew. Chem., Int. Ed. Engl., 2009, 48, 4816–4819 CrossRef CAS PubMed.
- I.-S. Cho, D. W. Kim, S. Lee, C. H. Kwak, S.-T. Bae, J. H. Noh, S. H. Yoon, H. S. Jung, D.-W. Kim and K. S. Hong, Adv. Funct. Mater., 2008, 18, 2154–2162 CrossRef CAS.
- F. Zhang, Z. Zhao, R. Tan, Y. Guo, L. Cao, L. Chen, J. Li, W. Xu, Y. Yang and W. Song, J. Colloid Interface Sci., 2012, 386, 277–284 CrossRef CAS PubMed.
- S. Tamura, N. Imanaka, M. Kamikawa and G. Adachi, Adv. Mater., 2000, 12, 898–901 CrossRef CAS.
- K. Ding, H. Gu, C. Zheng, L. Liu, L. Liu, X. Yan and Z. Guo, Electrochim. Acta, 2014, 146, 585–590 CrossRef CAS.
- M. D. Marcos, P. Amoros, A. Beltran-Porter, R. Martinez-Manez and J. P. Attfield, Chem. Mater., 1993, 5, 121–128 CrossRef CAS.
- X. Liu, Y. Xing, X. Wang, H. Xu, X. Liu, K. Shao and Z. Su, Chem. Commun., 2010, 46, 2614–2616 RSC.
- Z. Gu, T. Zhai, B. Gao, G. Zhang, D. Ke, Y. Ma and J. Yao, Cryst. Growth Des., 2007, 7, 825–830 CAS.
- Y. Ni, K. Liao, J. Hong and X. Wei, CrystEngComm, 2009, 11, 570–575 RSC.
- H. Pang, S. Wang, W. Shao, S. Zhao, B. Yan, X. Li, S. Li, J. Chen and W. Du, Nanoscale, 2013, 5, 5752–5757 RSC.
- J. Zhao, H. Pang, J. Deng, Y. Ma, B. Yan, X. Li, S. Li, J. Chen and W. Wang, CrystEngComm, 2013, 15, 5950–5955 RSC.
- X. Sun, Y. Xing, X. Liu, B. Liu and S. Hou, Dalton Trans., 2010, 39, 1985–1988 RSC.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- I. E. Rauda, V. Augustyn, B. Dunn and S. H. Tolbert, Acc. Chem. Res., 2013, 46, 1113–1124 CrossRef CAS PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- X. Lu, M. Yu, G. Wang, Y. Tong and Y. Li, Energy Environ. Sci., 2014, 7, 2160–2181 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- Y. Shi, L. Peng, Y. Ding, Y. Zhao and G. Yu, Chem. Soc. Rev., 2015, 44, 6684–6696 RSC.
- L. Li, S. Peng, H. B. Wu, L. Yu, S. Madhavi and X. W. D. Lou, Adv. Energy Mater., 2015, 5, 1500753 CrossRef.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
- S. Peng, L. Li, H. B. Wu, S. Madhavi and X. W. Lou, Adv. Energy Mater., 2014, 1401172 Search PubMed.
- H. Xu, X. Hu, H. Yang, Y. Sun, C. Hu and Y. Huang, Adv. Energy Mater., 2014, 1401882 Search PubMed.
- X. Lu, T. Zhai, X. Zhang, Y. Shen, L. Yuan, B. Hu, L. Gong, J. Chen, Y. Gao, J. Zhou, Y. Tong and Z. L. Wang, Adv. Mater., 2012, 24, 938–944 CrossRef CAS PubMed.
- H. Pang, Y. Liu, J. Li, Y. Ma, G. Li, Y. Ai, J. Chen, J. Zhang and H. Zheng, Nanoscale, 2013, 5, 503–507 RSC.
- H. Pang, C. Wei, Y. Ma, S. Zhao, G. Li, J. Zhang, J. Chen and S. Li, ChemPlusChem, 2013, 78, 546–553 CrossRef CAS.
- J. Zhao, S. Wang, Z. Run, G. Zhang, W. Du and H. Pang, Part. Part. Syst. Charact., 2015, 32, 880–885 CrossRef CAS.
- H. B. Li, M. H. Yu, F. X. Wang, P. Liu, Y. Liang, J. Xiao, C. X. Wang, Y. X. Tong and G. W. Yang, Nat. Commun., 2013, 4, 1894 CrossRef CAS PubMed.
- Y. Xi, B. Dong, Y. Dong, N. Mao, L. Ding, L. Shi, R. Gao, W. Liu, G. Su and L. Cao, Chem. Mater., 2016, 28, 1355–1362 CrossRef CAS.
- C. Hao, F. Wen, J. Xiang, L. Wang, H. Hou, Z. Su, W. Hu and Z. Liu, Adv. Funct. Mater., 2014, 24, 6700–6707 CrossRef CAS.
- X. Cao, B. Zheng, W. Shi, J. Yang, Z. Fan, Z. Luo, X. Rui, B. Chen, Q. Yan and H. Zhang, Adv. Mater., 2015, 27, 4695–4701 CrossRef CAS PubMed.
- X. Xia, J. Tu, Y. Zhang, X. Wang, C. Gu, X. B. Zhao and H. J. Fan, ACS Nano, 2012, 6, 5531–5538 CrossRef CAS PubMed.
- J. Zhang and X. S. Zhao, ChemSusChem, 2012, 5, 818–841 CrossRef CAS PubMed.
- S. Baskar, Z. Khan, S. Park, K. Kim, H. Ko and Y. Kim, J. Mater. Chem. A, 2015, 3, 21553–21561 Search PubMed.
- X.-F. Lu, D.-J. Wu, R.-Z. Li, Q. Li, S.-H. Ye, Y.-X. Tong and G.-R. Li, J. Mater. Chem. A, 2014, 2, 4706–4713 CAS.
- R. Li, X. Tao and X. Li, J. Mater. Chem., 2009, 19, 983–987 RSC.
- S. Nayak, B. Sahoo, T. K. Chaki and D. Khastgir, RSC Adv., 2014, 4, 1212–1224 RSC.
- M. Prabu and K. Ramalingam, RSC Adv., 2015, 5, 18554–18564 RSC.
- C. Wei, C. Cheng, S. Wang, Y. Xu, J. Wang and H. Pang, Chem.–Asian J., 2015, 10, 1731–1737 CrossRef CAS PubMed.
- A. M. Lazarin and C. Airoldi, Anal. Chim. Acta, 2004, 523, 89–95 CrossRef CAS.
- K. Sugiyama and M. Tokonami, Mineral. J., 1990, 15, 141–146 CrossRef CAS.
- S. Zhai, C.-C. Lin and W. Xue, Vib. Spectrosc., 2014, 70, 6–11 CrossRef CAS.
- G. Gao, H. B. Wu, S. Ding, L. M. Liu and X. W. Lou, Small, 2015, 11, 804–808 CrossRef CAS PubMed.
- K.-W. Nam and K.-B. Kim, J. Electrochem. Soc., 2002, 149, A346–A354 CrossRef CAS.
- B. Wang, J. S. Chen, Z. Wang, S. Madhavi and X. W. D. Lou, Adv. Energy Mater., 2012, 2, 1188–1192 CrossRef CAS.
- S. D. Perera, X. Ding, A. Bhargava, R. Hovden, A. Nelson, L. F. Kourkoutis and R. D. Robinson, Chem. Mater., 2015, 27, 7861–7873 CrossRef CAS.
- S. Guo, J. Liu, S. Qiu, W. Liu, Y. Wang, N. Wu, J. Guo and Z. Guo, J. Mater. Chem. A, 2015, 3, 23895–23904 CAS.
- Y. Wang, H. Wei, J. Wang, J. Liu, J. Guo, X. Zhang, B. L. Weeks, T. D. Shen, S. Wei and Z. Guo, J. Mater. Chem. A, 2015, 3, 20778–20790 CAS.
- H. Li, Y. Gao, C. Wang and G. Yang, Adv. Energy Mater., 2015, 5, 1401767 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08149e |
| This journal is © The Royal Society of Chemistry 2016 |