DOI:
10.1039/C6RA08053G
(Paper)
RSC Adv., 2016,
6, 43771-43779
Site-preferential occupancy induced photoluminescence tuning in (Ca,Ba)5(PO4)3Cl:Eu2+ phosphors†
Received
29th March 2016
, Accepted 22nd April 2016
First published on 25th April 2016
Abstract
Apatite structured (Ca1−xBax)5(PO4)3Cl:Eu2+ (0 ≤ x ≤ 1) solid-solution phosphors were successfully prepared via a typical Pechini sol–gel method. Structural refinement confirms the formation of solid-solution phases under the whole substitution ratio. All samples crystallized in a hexagonal phase with a space group of P63/m (176), and there are two kinds of cation sites (4f and 6h) in the host lattice. The as-prepared (Ca1−xBax)5(PO4)3Cl:Eu2+ phosphors show similar broad absorptions from 250 nm to 450 nm with peaks around 397 nm. Under 397 nm UV, Ca5(PO4)3Cl:Eu2+ presents a blue emission centered at 460 nm. By substituting Ba2+ for Ca2+, an unexpected red-shift up to 490 nm was first observed at x ≤ 0.5 and then a blue-shift after x > 0.5 occurred with a resulting emission at 438 nm for x = 1. The abnormal red-shift and blue-shift were revealed by the Rietveld structural refinement method and the possible luminescence mechanisms were proposed. The former is attributed to the preferential occupancy of 6h sites by Eu2+ ions and the expansion of neighboring Ba/Ca–O bond lengths. The latter mainly results from the entering of Eu2+ ions into the looser Ba2+ sites. The proposed luminescence mechanism can help reveal the underlying mechanisms in optical adjustment by changing the coordination environment at local sites. In addition, the thermal stability of (Ca,Ba)5(PO4)3Cl:Eu2+ phosphors were systematically investigated. Generally, the as-prepared (Ca,Ba)5(PO4)3Cl:Eu2+ phosphors can act as potential emitting-tunable phosphors for potential applications in n-UV based white LEDs.
1. Introduction
In recent years, InGaN-based white light-emitting diodes (WLEDs) have attracted substantial attention for application in display lighting sources and solid-state illuminating systems due to their high energy efficiency, low energy consumption, reliability, durability, long lifetimes, and ecofriendly features.1–3 Phosphor materials are important in generating high quality white lighting sources in WLEDs devices. A recent surge of research on phosphor-converted WLEDs has given researchers new opportunities and challenges in exploring new and highly efficient phosphors.4–9 To date, many studies have devoted to develop strategies for designing novel phosphor materials and optimizing their luminescence properties including single crystal growth method,10 cationic/anionic substitutions,11–19 solid state combinatorial chemistry method,20 single-particle-diagnosis approach,21 design of energy transfers at different sites,22–27 and so on. These approaches commonly change the coordination environment surrounding activators (especially Ce3+ and Eu2+ ions) because the 5d–4f transitions are sensitive to structural variation.3,7 Especially, various cation substitutions have been widely used to adjust luminescent properties of phosphor materials, and lots of luminescent mechanisms have been proposed to clarify the dependence of structure and luminescence such as ‘Cation-Size-Mismatch effect’,13 ‘Local shrinkage effect’,28 ‘Neighboring-Cation Substitution effect’,28 ‘Chemical Unit Cosubstitution’,11 ‘Nanosegregation and Neighbor-Cation Control effect’,12,14 ‘Mixing of nanophases’12,14,28 and so on. These photoluminescence tuning strategies could be used to tune spectral position and shape, and improve the luminescence efficiency and thermal stability of phosphors materials, which promotes their application in phosphor-converted WLEDs.9,10,29 However, these mechanisms could not solve all the problems once and for all in phosphors, and there are still some underlying mechanisms unclear and need be clarified for further tuning and optimizing the luminescence performances of the current phosphors.
Rare-earth activated chlorapaptite luminescence materials have been widely reported because of a facile synthesis, low cost, excellent optical properties and highly physical-chemical stability.30–41 In this structures, it typically contains the nine-coordinated 4f sites with C3 point symmetry and the eight-coordinated 6h sites with Cs point symmetry.32,38 Moreover, the coordination environment of 4f and 6h sites is easily effected by compositional substitution, generating tunable luminescence properties.42–48 Some cation-substitution strategies have also been used in the Ce3+ or Eu2+-doped (Ca, Sr, Ba)5(PO4)3Cl systems.32,37,38 However, there is no report to systematically clarify the abnormal redshift and blueshift luminescence phenomenon in Eu2+-doped (Ca, Sr, Ba)5(PO4)3Cl. Herein, we successfully prepared a series of Eu2+-doped (Ca,Ba)5(PO4)3Cl solid-solution phosphors via a Penchini sol–gel process. The present work focus on analyzing the structural variation of Ca5(PO4)3Cl:Eu2+ when gradually substituting Ba2+ for Ca2+ by Rietveld structural refinement. Then the dependence of structural variation and the abnormal redshift and blueshift luminescence was revealed and a possible luminescence mechanism was proposed. Finally, the thermal stabilities of (Ca,Ba)5(PO4)3Cl:Eu2+ phosphors were also systematically investigated.
2. Experimental
Chemicals and materials
CaCl2·2H2O (A.R.), BaCl2·2H2O (A.R.), NH4H2PO4 (A.R.), HCl (A.R.), citric acid and polyethylene glycol (PEG, molecular weight = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai). Eu2O3 (≥99.99%) were purchased from Science and Technology Parent Company of Changchun Institute of Applied Chemistry. All chemicals were of analytical grade reagents and used directly without further purification.
000) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai). Eu2O3 (≥99.99%) were purchased from Science and Technology Parent Company of Changchun Institute of Applied Chemistry. All chemicals were of analytical grade reagents and used directly without further purification.
Preparation
The Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) phosphor powders were synthesized by a Pechini sol–gel process.49 The doping concentration of Eu2+ ions was fixed at 2 mol% of the total content of (Ca2+/Ba2+) ions in the host. The as-prepared samples were marked as CPOCl, CPOCl–Ba0.25, CPOCl–Ba0.5, CPOCl–Ba0.75, BPOCl, respectively. Typically, stoichiometric Eu2O3 was firstly dissolved in dilute hydrochloric acid (HCl) under stirring and heating, and then stoichiometric amounts of CaCl2·2H2O, BaCl2·2H2O, and NH4H2PO4 were added. Subsequently, citric acid and polyethylene glycol (PEG, molecular weight = 20000) were dissolved in the above solution (CPEG = 0.01 M, citric acid:metal ion = 2:1 in per mole). The resultant mixtures were stirred for 1 h and heated in a 75 °C water bath until homogeneous gels formed. After being dried in an oven at 100 °C for 24 h, the gels were ground and prefired at 700 °C for 4 h in air. Next, the samples were fully ground and collected in the aluminum oxide crucibles, and then calcined in a horizontal tube furnace at 1250 °C for 4 h with a N2/H2 = 92%/8% atmosphere. After the furnace slowly cooled to room temperature, the sintered products were ground again, yielding the resulting phosphor powders.
Characterization
The finely ground powders were used in all measurements. The crystal structure and phase purity of the as-prepared samples were characterized by X-ray powder diffractometer (XRD), which were performed on a D8 Focus diffractometer (Bruker, Kalsruhe, Germany) at as canning rate of 1° min−1 in the 2θ range from 5° to 120° with Ni-filtered Cu-Kα (λ = 1.540598 Å). XRD Rietveld profile refinements of the structural models and texture analysis were performed with the use of General Structure Analysis System (GSAS) software. The morphologies of the samples were inspected using a field emission scanning electron microscope (FE-SEM, S-4800, Hitachi). The photoluminescence excitation (PLE) and emission (PL) spectra were measured by fluorescence spectrometer (Fluoromax-4P, Horiba Jobin Yvon, New Jersey, U.S.A.) equipped with a 450 W xenon lamp as the excitation source, and both excitation and emission spectra were recorded in 1.0 nm interval with the width of the monochromator slits adjusted as 0.50 nm. The absorption spectra were measured by UV-Visible diffuse reflectance spectroscopy UV-2550PC (Shimadzu Corporation, Japan). X-ray photoelectron spectroscopy (XPS) measurements were carried out on Mg Kα radiation source (Kratos XSAM-800 spectrometer). The thermal stability of luminescence of phosphor materials were measured by Fluoromax-4P spectrometer connected a heating equipment (TAP-02), and the samples were heated from 25 °C to 250 °C with a 25 °C interval. Before recording the emission spectra, each sample remained two minutes at the designed temperature. The luminescence decay curves were obtained from a Lecroy Wave Runner 6100 digital oscilloscope (1 GHz) using a tunable laser (pulse width = 4 ns, gate = 50 ns) as excitation source (Continuum Sunlite OPO). The photoluminescence quantum yields (QYs) were measured by absolute PL quantum yield measurement system C9920-02 (Hamamatsu photonics K.K., Japan).
3. Results and discussion
The phase purity of the studied samples were firstly confirmed by powder XRD patterns. Fig. 1 show the XRD patterns of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples. Obviously, the diffraction peaks of both ends of the samples, Ca4.9Eu0.1(PO4)3Cl (CPOCl) and Ba4.9Eu0.1(PO4)3Cl (BPOCl), could be well assigned to the pure hexagonal Ca5(PO4)3Cl phase (JCPDS #33-0271) and pure Ba5(PO4)3Cl phase (JCPDS #16-0686), respectively.38,47 When gradually substituting Ba2+ for Ca2+ in CPOCl, all XRD diffraction peaks of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0.25, 0.5, 0.75, 1) samples show linear shifts toward small-angle direction with the increase of Ba2+ content (x). This phenomenon is ascribed to the larger radius of Ba2+ ions (1.42 Å, CN = 8; 1.47 Å, CN = 9) than Ca2+ ions (1.12 Å, CN = 8; 1.18 Å, CN = 9), which means the successful incorporation of Ba2+ ions into the host lattice and the formation of solid-solutions according to the Vegard rule.29 Moreover, the whole solid-solution series all crystallized in hexagonal phase with space group of P63/m (176), namely, presented typical apatite structures. In order to further confirm the formation of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) solid solutions and depict their crystal structures, the XRD patterns were used to perform the Rietveld refinement with GASA software. The crystal structure of Ba5(PO4)3Cl from the literature were used as the initial parameters.41
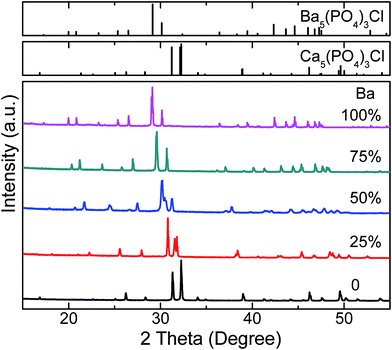 |
| | Fig. 1 XRD patterns of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples. The standard Ca5(PO4)3Cl data (JCPDS #33-0271) and Ba5(PO4)3Cl data (JCPDS #16-0686) are shown as references. | |
Fig. 2a presents the experimental, calculated, different XRD profiles and Bragg positions for the Rietveld refinement of the representative CPOCl–Ba0.5 sample at room temperature.
 |
| | Fig. 2 (a) Experimental (black crosses), calculated (red solid line) XRD patterns and their difference (blue solid line) for the Rietveld fit of Ca2.45Ba2.45Eu0.1(PO4)3Cl XRD pattern by the GSAS program. The short vertical lines show the positions of Bragg reflections of the calculated pattern. (b) Dependence of cell parameters (a, b, c), volume (V) and Ba doping concentration (x) in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl systems. (c) Schematic crystal structure diagrams for cation substitution at 4f and 6h sites of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.5, 1) samples. | |
According to the refinement result, the as-prepared CPOCl–Ba0.5 crystallizes in hexagonal phase with space group P63/m (176), a = b = 9.9150(4) Å, c = 7.2699(4) Å, α = β = 90°, γ = 120°, V = 618.93(5) Å3 and Z = 2. All atom positions, fraction factors, and thermal vibration parameters were refined by convergence and satisfied well the reflection conditions, Rwp = 5.58%, Rp = 3.65%, and χ2 = 3.331, as shown in Table 1. This result verifies that the formation of single-phase, and the crystal structure of apatite are unchanged with the introduction of Ba2+ ions. Other Rietveld refinement XRD patterns of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.75, 1) samples were shown in Fig. S1 (ESI†), and some main refined parameters of all samples were listed in Table 1. Clearly, the whole Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl series maintain the hexagonal phase structure. Smooth increases in lattice parameters (a, b, c, and V) with increasing x (Fig. 2b) also further confirm that the Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.75, 1) solid solutions were formed.11,12 According to the Rietveld refinement result, the spatial views of the representative Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.5, 1) unit cells are shown in Fig. 2c. They all show that Ca1 atoms located at 4f site coordinated with nine oxygen atoms to form tetrakaidecahedrons,47,50 and any two tetrakaidecahedrons are connected each other by sharing triangle plane, constructing a 4f site tunnel along c-axis. While the Ca2 atoms located at 6h site form hendecahedron with its surrounding six oxygen atoms and two chlorine atoms, and these hendecahedron connect each other through sharing edges. The neighboring tetrakaidecahedron and hendecahedron are connected through tetrahedral PO4 groups and sharing edges by each other. Although the studied samples present a similar crystal structure, there are different coordination environments around Ca2+/Ba2+ and Eu2+ ions including different bond lengths, bond angles and distortions of cation-O coordinated polyhedrons due to the mismatch of ion radius. The selected interatomic distances in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples were shown in Table S1 (ESI†). In addition, the SEM images of the representative CPOCl, CPOCl–Ba0.5 and BPOCl samples are shown in Fig. S2 (ESI†). It is found that the obtained samples all consisted of aggregated microcrystallines with smooth surface and particle sizes from 2 μm to 8 μm. Moreover, the morphologies and sizes are basically unchanged with the substitution of Ba2+ for Sr2+, implying a small effect of morphology and size on their luminescence properties in the studied system.
Table 1 Refined main structure parameters of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples derived from the GSAS refinement of XRD data
| Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl |
a = b, Å |
c, Å |
V, Å3 |
Rwp, % |
Rp, % |
χ2 |
| x = 0 |
9.60776(9) |
6.79318(8) |
543.061(10) |
7.03 |
4.11 |
4.368 |
| x = 0.25 |
9.74465(19) |
6.96423(14) |
572.712(20) |
5.02 |
3.23 |
2.747 |
| x = 0.50 |
9.9150(4) |
7.2699(4) |
618.93(5) |
5.58 |
3.65 |
3.331 |
| x = 0.75 |
10.0651999(0) |
7.4400001(0) |
652.7522583(0) |
5.22 |
3.35 |
2.615 |
| x = 1 |
10.25518(6) |
7.63833(5) |
695.690(8) |
5.59 |
3.63 |
3.096 |
It is known that Eu2+ ions activated phosphors have been widely used in solid state lighting area. Because the unprotected outer-shell electrons, the 5d energy levels of Eu2+ are strongly affected by the surrounding crystal field, symmetry, anion polarizability, and covalency of the host crystal.51–55 Fig. 3 presents the absorption spectra and the normalized photoluminescence excitation (PLE), photoluminescence (PL) of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples. Typically, the as-prepared samples all shows two broad-band absorption from 240 nm to 450 nm. The weak one (240–275 nm) is attributed to the host absorption, while the strong one from 275 nm to 450 nm centered around 390 nm is due to the electronic transitions from the 4f ground state of Eu2+ (8S7/2) to its 5d excited state.55 The excitation spectra of the studied samples are consistent with their absorption spectra except for the maximum excitation wavelength around 397 nm. Under 397 nm UV, CPOCl exhibits an asymmetric emission band ranging from 420 to 520 nm with the peak at 460 nm, which implies possible spectral overlaps coming from different luminescence centers. This should be attributed to two available Ca sites (Ca1, 4f sites; Ca2, 6h sites) in Ca5(PO4)3Cl host. Fig. S3 (ESI†) shows that the PL spectrum of CPOCl can be well-fitted with a sum of two Gaussian bands with peaks at 460 and 480 nm, which originates from Eu1 and Eu2 emission centers, respectively. Generally, the crystal field splitting (Dq) can be determined by the following equation:56,57
| |
 | (1) |
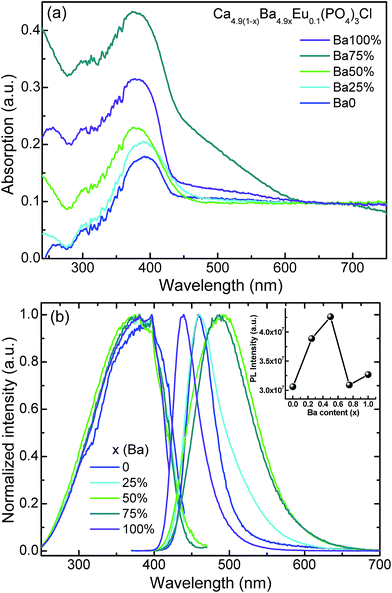 |
| | Fig. 3 (a) The absorption spectra and (b) the normalized photoluminescence excitation (PLE) and photoluminescence (PL) spectra of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples. The insert in (b) presents the PL intensity of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl as a function of Ba doping concentration (x). | |
where Dq is the magnitude of the 5d energy level separation, Z is the anion charge or valence, e is the electron charge, r is the radius of the d wavefunction, and R is the bond length. This equation can be used as an approximation for describing crystal field splitting trends with bond length.53 The shorter the bond length and the stronger the crystal field splitting, and then the longer emission wavelength will appear.44 According to refinement result of CPOCl (Table S1, (ESI†)), the average bond length rCa1–O and rCa2–O/Cl is 2.624 Å and 2.618 Å, respectively, indicating the crystal field strength is Dq (Ca1 sites) < Dq (Ca2 sites). Therefore, the Eu1 (high energy) and Eu2 (low energy) emission should be assigned to Ca1 (4f) and Ca2 (6h) sites, respectively. It is noted that the rCa1–O and rCa2–O/Cl in CPOCl sample is very close, which is consistent with its slightly asymmetric emission profile. However, an expected blueshifted emission with the substitution of Ba2+ ions for Ca2+ ions in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) systems was not observed, although there was a continuous expansion of crystal lattice in the studied samples (Fig. 2b). When gradually increasing the concentration of doped Ba2+ ions, the emission peaks of phosphors were firstly redshifted, reaching a maximum emission at 490 nm with x = 0.5, and then were blueshifted to 438 nm at x = 1. Therefore, their emission colors were tuned from blue light (0.1497, 0.1052) to green light (0.1799, 0.3175) and then to blue light (0.1529, 0.0476) again, which can be confirmed by the emission peaks, corresponding CIE color chromaticity coordinates and luminescence photos in Table 2 and Fig. 4. Similarly, the FWHM of their PL spectra were also firstly widened from 43 nm (x = 0) to 100 nm (x = 0.5) and finally narrowed to 41 nm (x = 1), as shown in Table 2. The possible reason is that the size-mismatch of Ba2+ and Ca2+ ions forms more cation sites with some distortions, which leads to more Eu2+ emission centers with different crystal field strengths. In addition, the as-prepared Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) sample present high quantum yields from 0.58 to 0.78 with x and it could be further improved by process optimization, which is beneficial to practical application in UV-based WLEDs lighting. The 4f–4f characteristic transitions of Eu3+ ions was not observed from the PL spectra of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples, indicating that the Eu3+ ions were almost completely reduced to Eu2+ ions. In order to further detect the chemical state of Eu in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples, the high resolution Eu 3d XPS analysis with a range from 1110 eV to 1180 eV of the representative CPOCl, CPOCl–Ba0.5, BPOCl samples are shown in Fig. S4 (ESI†). Obviously, the three samples showed the same peaks around the binding energy 1127 eV, which should be attributed to Eu2+ ions. However, the XPS peaks of Eu3+ ions around 1134 eV were not detected. The result further confirmed that the Eu3+ ions were completely reduced to Eu2+ ions in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples.
Table 2 The CIE chromaticity coordinates (X, Y), emission peaks, FWHM, and quantum yields (QYs) of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples (λex = 397 nm)
| Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl |
CIE X |
CIE Y |
Peak/nm |
FWHM/nm |
QEs |
| x = 0 |
0.1497 |
0.1052 |
459 |
43 |
0.56 |
| x = 0.25 |
0.1551 |
0.1746 |
461 |
57 |
0.72 |
| x = 0.50 |
0.1799 |
0.3175 |
491 |
100 |
0.78 |
| x = 0.75 |
0.1816 |
0.3309 |
487 |
86 |
0.57 |
| x = 1 |
0.1529 |
0.0476 |
439 |
41 |
0.60 |
 |
| | Fig. 4 The CIE chromaticity coordinates diagram for Ca4.9(1−x)Ba4.9x(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples. The insets are corresponding digital photographs of samples under the excitation of 365 nm UV lamp. | |
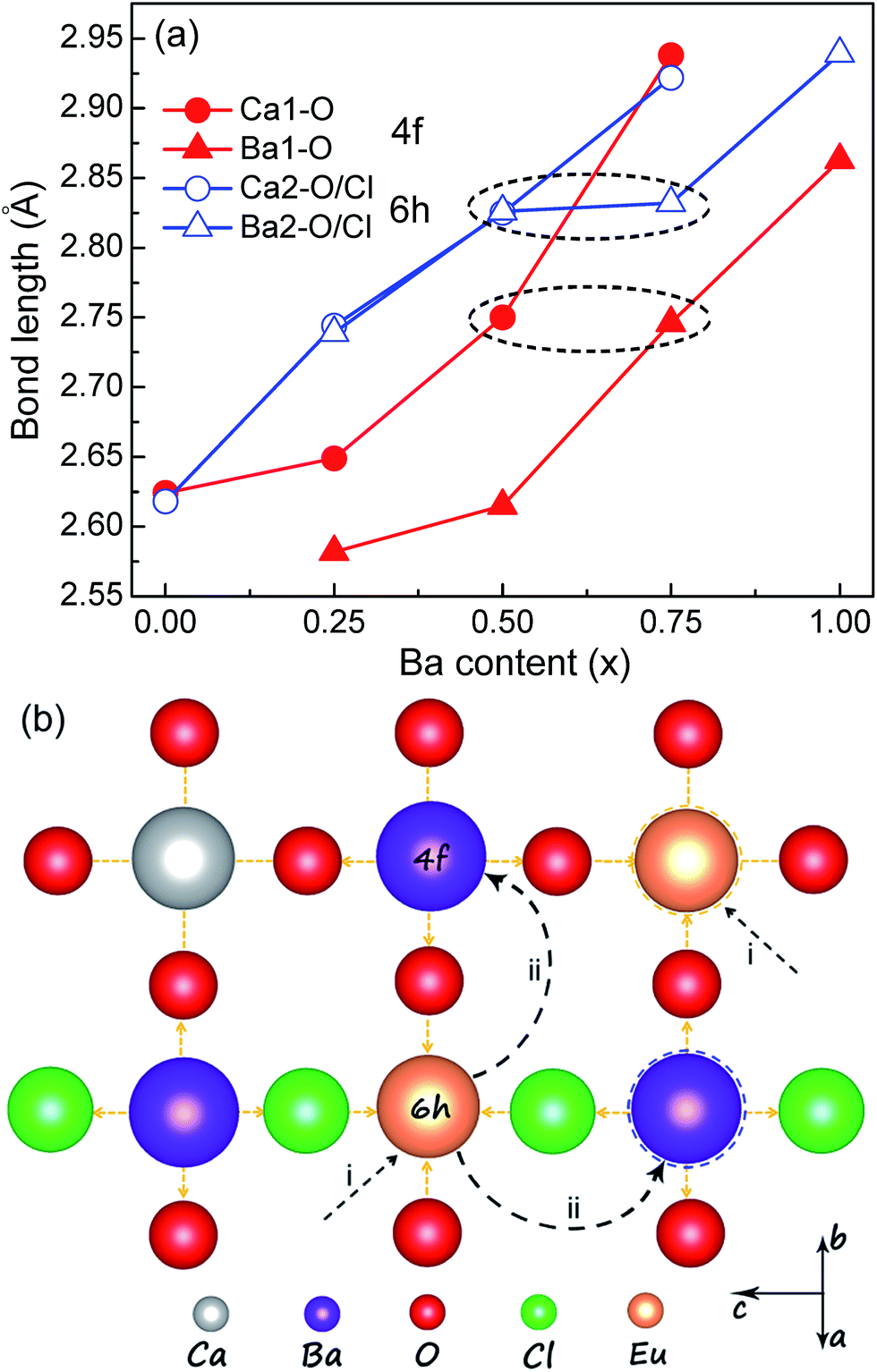
In order to clarify the abnormal redshift and blueshift mechanisms in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl systems, the Rietveld refinement method was used to reveal the local structural changes of Eu2+ ions. According to the refinement results, the blue emission in Ba4.9Eu0.1(PO4)3Cl is easily understood, which could be indexed to the preferential site occupancy of Eu2+ ions. Seen from Fig. 5a and Table S1 (ESI†), the average bond length rBa1–O (4f) and rBa2–O/Cl (6h) is 2.863 Å and 2.939 Å, respectively, which are obviously larger than that of rCa1–O and rCa2–O/Cl in CPOCl. This means that the crystal field strength is Dq (Ba1, 2) > Dq (Ca1, 2), and it is reasonable to observe the blueshifted emission of Eu2+ ions in BPOCl than that in CPOCl. Therefore, the blueshifted emission in BPOCl relative to that in CPOCl is due to the occupancy of the larger Ba2+ sites by Eu2+ ions (Fig. 5bii). It should be noted that the high-energy and low-energy emission of Eu2+ in BPOCl are respectively attributed to Ba2 (6h) and Ba1 (4f) sites, which is opposite to that of in CPOCl sample.
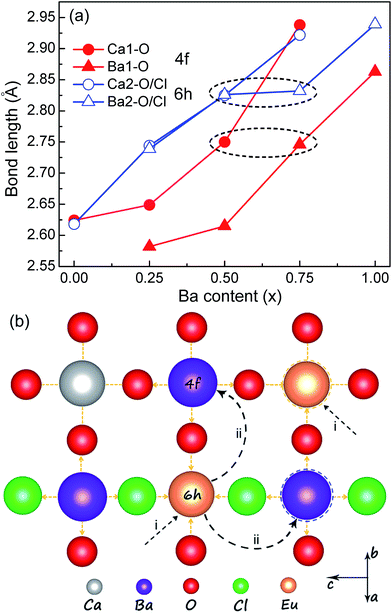 |
| | Fig. 5 (a) The variation of Ca–O and Ba–O bond lengths located at 4f and 6h sites in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) systems as a function of Ba doping contents (x). (b) A schematic explanation of red-shift and blue-shift luminescence mechanism in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) with x. | |
Except for the average structural variation, the change of local coordination environment around activator ions also has important influence on the luminescence properties of phosphors.11–14,28,29 Because the Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) solid solutions possess the same apatite structures (P63/m, 176), this cation-substitution induced abnormal redshifts from x = 0 to x = 0.5 samples possibly result from the change of local crystal lattice around Eu2+ ions. Because the Ca2+ sites dominates at x ≤ 0.5 in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5) samples, Eu2+ ions preferentially replace at Ca2+ sites. By analyzing the Gaussian fitting of the PL spectrum of CPOCl (Fig. S3, (ESI†)), the Eu1 emission at 4f sites are obviously stronger that that at 6h sites, indicating that Eu2+ ions mainly locate into Ca1 (4f) sites (Fig. 5bi). The close lifetime values (Eu1, 0.447 μs; Eu2, 0.426 μs) of CPOCl sample for different Eu2+ emission centers also confirm this result (Fig. 6). However, the increased ratio of rBa2–O/Cl/rBa1–O is larger than rCa2–O/Cl/rCa1–O with the doping of Ba2+ concentration to 0.25 and 0.50, as shown in Fig. 5a. This means that more Ba2+ ions gradually occupy more 6h sites with the increase of x. In addition, a widened full width at half maximum (FWHM) of the PL spectra for Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5) from 43 nm to 100 nm with the increase of x from 0 to 0.5 is observed in Fig. 3b and Table 2. This is because that the increase of Ba2+ concentration generates more cation sites to accommodate Eu2+ ions. These sites have different distortion degree due to the size-mismatch of Ba2+ and Ca2+ ions, creating more Eu2+ emission centers and finally increasing the FWHM. This result could be validated by the clearly different lifetime decay curves and values of CPOCl–Ba0.25 and CPOCl–Ba0.25 samples monitoring at different emission sites (Fig. 6b, c and f). Generally, although the bond lengths of Ca/Ba–O/Cl gradually increase with the increase of Ba2+-doping concentration in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5), the predicted blue shift are not appear. The available red shift should originate from the expansion effect of neighboring cations, which can be proposed as follows (Fig. 5b): first, Eu2+ preferentially occupy Ca2+ (4f and 6h) sites in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5). Then, the lattice strain of Ca–O bond increases when the neighboring Ca2+ ions are replaced by the larger Ba2+ ions. To release lattice strain, the Eu–O bond length decreases. With the increase of Ba2+ content, other neighboring Ca2+ sites are successively replaced by Ba2+ ions, thereby gradually decreasing the Eu–O bond length. This result enlarges the 5d-orbital crystal field splitting of Eu2+ ions, causing a continuous redshift in the emission spectra. Finally, it is found from Fig. 5a (black dash circle) and Table 1 that the rBa1–O and rBa2–O/Cl of CPOCl–Ba0.75 are basically equal to the rCa1–O and rCa2–O/Cl, respectively, implying the almost same crystal field environment of Ba2+ and Ca2+ ions. This could be intuitively confirmed by the close lifetimes 0.645 μs for Ca1 and 0.677 μs for Ba1 in CPOCl–Ba0.5 as well as 0.967 μs for Ca2 and 0.991 μs for Ba2 in CPOCl–Ba0.5, as shown in Fig. 6c, d, and f. While the emission spectrum of CPOCl–Ba0.75 is almost same to CPOCl–Ba0.5 except for a slightly narrower FWHM. Therefore, it could be concluded that Eu2+ ions were preferentially transferred to the Ba2+ sites from Ca2+ sites in view of the dominant Ba2+ sites in CPOCl–Ba0.75.
 |
| | Fig. 6 The luminescence lifetime decay curves of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples monitoring at different emission sites: (a) x = 0; (b) x = 0.25; (c) x = 0.5; (d) x = 0.75; (e) x = 1. (f) The fitting lifetimes values of the above samples. | |
The thermal stability of luminescence of phosphors is an important index to be considered in practical WLED lighting applications, which influences the lighting efficiency and quality.58–64 A comprehensive understanding of the thermal quenching of phosphors is indispensable because high-power LEDs suffer from thermal problem.62 Fig. 7a and b show the temperature-dependent PL spectra of the representative Ca2.45Ba2.45Eu0.1(PO4)3Cl sample and the dependence of emission intensity of the as-prepared Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) on temperatures from 25 °C to 250 °C, respectively. Generally, the emission intensity of phosphors decreases with increasing temperature because of nonradiative transitions from the excited state to the ground state. In this study, the as-prepared Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) present good thermal stability, which basically all maintain higher 80% of the room intensity at 150 °C. For example, the green emitting CPOCl–Ba0.5 can still only retain 86% of the original value at 150 °C.55 Moreover, there is no spectral shift in CPOCl–Ba0.5 with temperatures, implying an excellent color stability. This result can be more clearly seen from the normalized PL spectra from 4f and 6h sites in the insert of Fig. 7a. It is note that there are blueshifted and redshifted emission in CPOCl and BPOCl, respectively, as shown in Fig. S5 (ESI†). This originates from the higher thermal stability of Eu2+ at the looser sites relative to that at the tight sites, which is consistent with the previous report.47 Fig. 7b shows that the thermal stabilities of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) firstly decrease and then increase, which are closely related to their activation energy (ΔE). The ΔE could be calculated according to the following equation:47,60
| | |
IT = I0/[1 + cexp(−ΔE/kT)]
| (2) |
where
I0 is the initial PL intensity of the phosphor at room temperature,
IT is the PL intensity at other temperatures,
c is a constant, Δ
E is the activation energy for thermal quenching, and
k is the Boltzmann constant (8.62 × 10
−5 eV). The calculated Δ
E of Ca
4.9(1−x)Ba
4.9xEu
0.1(PO
4)
3Cl (
x = 0, 0.25, 0.5, 0.75, 1) are 0.363, 0.351, 0.324, 0.333, 0.366 eV, respectively. The relatively high activation energy results in good thermal stabilities for these phosphors. Generally, the activation energy increases with the decrease in Stokes shift, thereby suggesting that the probability of nonradiative transition can be weakened by thermal activation. In Ca
2.45Ba
2.45Eu
0.1(PO
4)
3Cl systems, the continuous redshifts at
x = 0–0.5 lead the decrease in Stokes shift. Therefore, the thermal quenching is CPOCl < CPOCl–Ba
0.25 < CPOCl–Ba
0.5. After
x > 0.50, their Stoke shifts have a continuous blueshift, and thus the activation energy gradually increase, leading to a decreasing thermal quenching, as shown the insert in
Fig. 7b.
 |
| | Fig. 7 (a) Normalized PL spectra of Ca2.45Ba2.45Eu0.1(PO4)3Cl (λex = 400 nm) at different temperatures (25–250 °C). (b) The dependence of emission intensity of Ca2.45Ba2.45Eu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) on temperatures from 25 °C to 250 °C. The inset in (b) shows the dependence of the activated energy (Ea) vs. Ba concentration (x). | |
4. Conclusion
In this work, (Ca,Ba)5(PO4)3Cl:Eu2+ solid solution phosphors were prepared by the Pechini sol–gel method. Smooth increase in cell parameters and volume with the substitution of Ba2+ ions for Ca2+ ions confirms the formation of solid solution phases. The as-prepared phosphors show a broad absorption from 250–450 nm with the maximum at 397 nm. Under 397 nm UV, the non-substituted Ca5(PO4)3Cl:Eu2+ exhibits a blue emission ranging from 420 to 520 nm with the peak at 460 nm. Interestingly, a first redshifted and then blueshifted emission was observed instead of an expected linear blueshifted emission. This abnormal redshift and blueshift mechanisms in the studied samples were revealed by the Rietveld refinement analysis of the variation of bond lengths. According to the refinement results, a possible redshift luminescence of doped Eu2+ ions at x ≤ 0.5 should be attributed to the firstly preferential occupancy of Ca sites by Eu2+ ions, and the subsequent shrinkage in Eu–O/Cl bond due to the gradual expansion of neighboring Ca/Ba–O/Cl bond length with the doping of Ba2+ ions. While the blueshift is mainly ascribed to the preferential occupancy of Ba2+ sites at x > 0.5. Therefore, a tunable emission from blue (0.1497, 0.1052) to green (0.1799, 0.3175), and then to blue (0.1529, 0.0476) could be realized in (Ca,Ba)5(PO4)3Cl:Eu2+ by simple cation-substitution. In addition, the as-prepared phosphors present good thermal stabilities (>80% of RT intensity at 150 °C) and high quantum yields (56–78%). The corresponding thermal quenching mechanism was also revealed. Generally, the as-prepared (Ca,Ba)5(PO4)3Cl:Eu2+ phosphors can act as potential color-tunable phosphors for possible applications in n-UV based white LEDs. The proposed photoluminescence tuning mechanism might be expected to work in other optical adjustment based on the local structural variation.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21301162, 51472234, 51172227, 91433110), National Basic Research Program of China (2014CB643803), Joint Funds of the National Natural Science Foundation of China (U1301242), the Fundamental Research Funds for National University, China University of Geosciences (Wuhan) (No. GBL31510), and the National College Students' Innovative Training Program (No. 201510491109, 201610491067, 201610491070).
Notes and references
- C. C. Lin and R. S. Liu, J. Phys. Chem. Lett., 2011, 2, 1268–1277 CrossRef CAS PubMed.
- V. Bachmann, C. Ronda, O. Oeckler, W. Schnick and A. Meijerink, Chem. Mater., 2009, 21, 316–325 CrossRef CAS.
- P. F. Smet, A. B. Parmentier and D. Poelman, J. Electrochem. Soc., 2011, 158, R37–R54 CrossRef CAS.
- W. B. Im, N. N. Fellows, S. P. DenBaars, R. Seshadri and Y.-I. Kim, Chem. Mater., 2009, 21, 2957–2966 CrossRef CAS.
- R. J. Xie and N. Hirosaki, Sci. Technol. Adv. Mater., 2007, 8, 588–600 CrossRef CAS.
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef CAS PubMed.
- N. C. George, K. A. Denault and R. Seshadri, Annu. Rev. Mater. Res., 2013, 43, 481–501 CrossRef CAS.
- P. Pust, A. Wochnik, E. Baumann, P. J. Schmidt, D. Wiechert, C. Scheu and W. Schnick, Chem. Mater., 2014, 26, 3544–3549 CrossRef CAS.
- F. W. Kang, X. B. Yang, M. Y. Peng, L. Wondraczek, Z. J. Ma, Q. Y. Zhang and J. R. Qiu, J. Phys. Chem. C, 2014, 118, 7515–7522 CAS.
- F. W. Kang, H. S. Zhang, L. Wondraczek, X. B. Yang, Y. Zhang, D. Y. Lei and M. Y. Peng, Chem. Mater., 2016, 28, 2692–2703 CrossRef CAS.
- Z. G. Xia, C. G. Ma, M. S. Molokeev, Q. L. Liu, K. Rickert and K. R. Poeppelmeier, J. Am. Chem. Soc., 2015, 137, 12494–12497 CrossRef CAS PubMed.
- Z. G. Xia, G. K. Liu, J. G. Wen, Z. G. Mei, M. Balasubramanian, M. S. Molokeev, L. C. Peng, L. Gu, D. J. Miller, Q. L. Liu and K. R. Poeppelmeier, J. Am. Chem. Soc., 2016, 138, 1158–1161 CrossRef CAS PubMed.
- W. T. Chen, H. S. Sheu, R. S. Liu and J. P. Attfield, J. Am. Chem. Soc., 2012, 134, 8022–8025 CrossRef CAS PubMed.
- W. Y. Huang, F. Yoshimura, K. Ueda, Y. Shimomura, H. S. Sheu, T. S. Chan, H. F. Greer, W. Z. Zhou, S. F. Hu, R. S. Liu and J. P. Attfield, Angew. Chem., Int. Ed., 2013, 52, 8102–8106 CrossRef CAS PubMed.
- K. A. Denault, J. Brgoch, M. W. Gaultois, A. Mikhailovsky, R. Petry, H. Winkler, S. P. DenBaars and R. Seshadri, Chem. Mater., 2014, 26, 2275–2282 CrossRef CAS.
- Y. Sato, H. Kato, M. Kobayashi, T. Masaki, D. H. Yoon and M. Kakihana, Angew. Chem., Int. Ed., 2014, 53, 7756–7759 CrossRef CAS PubMed.
- Z. Y. Zhao, Z. G. Yang, Y. R. Shi, C. Wang, B. T. Liu, G. Zhu and Y. H. Wang, J. Mater. Chem. C, 2013, 1, 1407–1412 RSC.
- N. Komuro, M. Mikami, Y. Shimomura, E. G. Bithellc and A. K. Cheethamd, J. Mater. Chem. C, 2015, 3, 204–210 RSC.
- A. Kalaji, M. Mikami and A. K. Cheetham, Chem. Mater., 2014, 26, 3966–3975 CrossRef CAS.
- W. B. Park, S. P. Singh and K. S. Sohn, J. Am. Chem. Soc., 2014, 136, 2363–2373 CrossRef CAS PubMed.
- N. Hirosaki, T. Takeda, S. Funahashi and R. J. Xie, Chem. Mater., 2014, 26, 4280–4288 CrossRef CAS.
- C. H. Huang, T. W. Kuo and T. M. Chen, ACS Appl. Mater. Interfaces, 2010, 2, 1395–1399 CAS.
- W. R. Liu, C. H. Huang, C. W. Yeh, Y. C. Chiu, Y. T. Yeh and R. S. Liu, RSC Adv., 2013, 3, 9023–9028 RSC.
- N. Guo, Y. J. Huang, H. P. You, M. Yang, Y. H. Song, K. Liu and Y. H. Zheng, Inorg. Chem., 2010, 49, 10907–10913 CrossRef CAS PubMed.
- Y. F. Liu, X. Zhang, Z. D. Hao, X. J. Wang and J. H. Zhang, J. Mater. Chem., 2011, 21, 6354–6358 RSC.
- X. Yu, T. Wang, X. H. Xu, T. M. Jiang, H. L. Yu, Q. Jiao, D. C. Zhou and J. B. Qiu, RSC Adv., 2014, 4, 963–968 RSC.
- D. G. Deng, H. Yu, Y. Q. Li, Y. J. Hua, G. H. Jia, S. L. Zhao, H. P. Wang, L. H. Huang, Y. Y. Li, C. X. Li and S. Q. Xu, J. Mater. Chem. C, 2013, 1, 3194–3199 RSC.
- G. G. Li, C. C. Lin, W. T. Chen, M. S. Molokeev, V. V. Atuchin, C. Y. Chiang, W. Z. Zhou, C. W. Wang, W. S. Li, H. S. Sheu, T. S. Chan, C. G. Ma and R. S. Liu, Chem. Mater., 2014, 26, 2991–3001 CrossRef CAS.
- M. Y. Peng, X. W. Yin, P. A. Tanner, M. G. Brik and P. F. Li, Chem. Mater., 2015, 27, 2938–2945 CrossRef.
- S. S. Wang, W. T. Che, Y. Li, J. Wang, H. S. Sheu and R. S. Liu, J. Am. Chem. Soc., 2013, 135, 12504–12507 CrossRef CAS PubMed.
- D. Y. Zhai, L. X. Ning, Y. C. Huang and G. K. Liu, J. Phys. Chem. C, 2014, 118, 16051–16059 CAS.
- R. J. Yu, C. F. Guo, T. Li and Y. Xu, Curr. Appl. Phys., 2013, 13, 880–884 CrossRef.
- Z. J. Wang, P. L. Li, Z. P. Yang and Q. L. Guo, J. Lumin., 2014, 151, 170–175 CrossRef CAS.
- P. L. Li, Z. J. Wang, Z. P. Yang and Q. L. Guo, RSC Adv., 2014, 4, 27708–27713 RSC.
- P. L. Li, Z. J. Wang, Z. P. Yang and Q. L. Guo, J. Mater. Chem. C, 2014, 2, 7823–7829 RSC.
- M. H. Hwang, E. Y. Lee, S. H. Hong, Y. B. Sun and Y. J. Kim, J. Electrochem. Soc., 2009, 156, J185–J188 CrossRef CAS.
- X. G. Zhang, J. L. Zhang, J. Q. Huang, X. P. Tang and M. L. Gong, J. Lumin., 2010, 130, 554–559 CrossRef CAS.
- W. N. Wang, F. Iskandar, K. Okuyama and Y. Shinomiya, Adv. Mater., 2008, 20, 3422–3426 CrossRef CAS.
- J. Yu, C. C. Guo, Z. Y. Ren and J. T. Bai, Opt. Laser Technol., 2011, 43, 762–766 CrossRef CAS.
- H. Y. Zou, Y. H. Song, Y. F. Deng, H. G. Zhang, Y. Sheng, K. Y. Zheng, X. Q. Zhou and J. Chen, J. Nanopart. Res., 2013, 15, 1973–1982 CrossRef.
- Z. Zhang, J. Wang, M. Zhang and Q. Su, Appl. Phys. B, 2008, 91, 529–537 CrossRef CAS.
- Z. G. Xia, M. S. Molokeev, W. B. Im, S. Unithrattil and Q. L. Liu, J. Phys. Chem. C, 2015, 119, 9488–9495 CAS.
- Q. Peng, C. M. Liu, D. J. Hou, W. J. Zhou, C. G. Ma, G. K. Liu, M. G. Brik, Y. Tao and H. B. Liang, J. Phys. Chem. C, 2016, 120, 569–580 CAS.
- Y. F. Xia, J. Chen, Y. G. Liu, M. S. Molokeev, M. Guan, Z. H. Huang and M. H. Fang, Dalton Trans., 2016, 45, 1007–1015 RSC.
- G. S. R. Raju, J. Y. Park, H. C. Jung, B. K. Moon, J. H. Jeong and J. H. Kim, J. Electrochem. Soc., 2011, 158, J20–J26 CrossRef CAS.
- Z. X. Tao, Y. L. Huang and H. J. Seo, Dalton Trans., 2013, 42, 2121–2129 RSC.
- Y. Tian, Y. Wei, Y. Zhao, Z. W. Quan, G. G. Li and J. Lin, J. Mater. Chem. C, 2016, 4, 1281–1294 RSC.
- P. P. Dai, C. Li, X. T. Zhang, J. Xu, X. Chen, X. L. Wang, Y. Jia, X. J. Wang and Y. C. Liu, Light: Sci. Appl., 2016, 5, e16024 CrossRef CAS.
- M. M. Shang, J. Fan, H. Z. Lian, Y. Zhang, D. L. Geng and J. Lin, Inorg. Chem., 2014, 53, 7748–7755 CrossRef CAS PubMed.
- Y. Q. Shen, R. Chen, F. Xiao, H. D. Sun, A. Tok and Z. L. Dong, J. Solid State Chem., 2010, 183, 3093–3099 CrossRef CAS.
- C. Wang, Z. Y. Zhao, Q. S. Wu, G. Zhu and Y. H. Wang, Dalton Trans., 2015, 44, 10321–10329 RSC.
- K.-S. Sohn, B. Lee, R.-J. Xie and N. Hirosaki, Opt. Lett., 2009, 34, 3427–3430 CrossRef CAS PubMed.
- Y. Shimomura, T. Honma, M. Shigeiwa, T. Akai, K. Okamoto and N. Kijima, J. Electrochem. Soc., 2007, 154, J35–J38 CrossRef CAS.
- M. Seibald, T. Rosenthal, O. Oeckler and W. Schnick, Crit. Rev. Solid State Mater. Sci., 2014, 39, 215–229 CrossRef CAS.
- W. B. Im, Y. Kim, H. S. Yoo and D. Y. Jeon, Chem. Mater., 2009, 48, 557–564 CAS.
- P. Dorenbos, ECS Solid State Lett., 2013, 2, R3001–R3011 CrossRef CAS.
- C. K. Jørgensen, Modern Aspects of Ligand Field Theory, Amsterdam: North-Holland, 1971 Search PubMed.
- X. Piao, K. Machida, T. Horikawa, H. Hanzawa, Y. Shimomura and N. Kijima, Chem. Mater., 2007, 19, 4592–4599 CrossRef CAS.
- W. B. Im, N. George, J. Kurzman, S. Brinkley, A. Mikhailovsky, J. Hu, B. F. Chmelka, S. P. DenBaars and R. Seshadri, Adv. Mater., 2011, 23, 2300–2305 CrossRef CAS PubMed.
- C. W. Yeh, W. T. Chen, R. S. Liu, S. F. Hu, H. S. Sheu, J. M. Chen and H. T. Hintzen, J. Am. Chem. Soc., 2012, 134, 14108–14117 CrossRef CAS PubMed.
- W. P. Chen, Dalton Trans., 2015, 44, 17730–17735 RSC.
- N. Komuro, M. Mikami, Y. Shimomura, E. G. Bithellc and A. K. Cheethamd, J. Mater. Chem. C, 2014, 2, 6084–6089 RSC.
- Y. Q. Li, J. E. J. van Steen, J. W. H. van Krevel, G. Botty, A. C. A. Delsing, F. J. DiSalvo, G. de With and H. T. Hintzen, J. Alloys Compd., 2006, 417, 273–279 CrossRef CAS.
- S. Y. Zhang, Y. Nakai, T. Tsuboi, Y. L. Huang and H. J. Seo, Chem. Mater., 2011, 23, 1216–1224 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The selected interatomic distances in Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples (Table S1); the Rietveld fitting of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.75, 1) XRD patterns (Fig. S1); the SEM image of the representative CPOCl, CPOCl–Ba0.5, BPOCl samples (Fig. S2); the normalized Gaussian peaks fitting photoluminescence emission (PL) spectra of Ca4.9(1−x)Ba4.9xEu0.1(PO4)3Cl (x = 0, 0.25, 0.5, 0.75, 1) samples at 4f sites and 6h sites (Fig. S3); the high resolution Eu 3d XPS spectra for the representative CPOCl, CPOCl–Ba0.5, BPOCl samples (Fig. S4); the normalized PL spectra of Ca2.45Ba2.45Eu0.1(PO4)3Cl (x = 0, 0.25, 0.75, 1) with temperatures from 25 °C to 250 °C (Fig. S5). See DOI: 10.1039/c6ra08053g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.