
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-responsive multicompartment nanofibers for controlled release of payloads†
Shuai
Jiang
abc,
Li-Ping
Lv
ad,
Katharina
Landfester
a and
Daniel
Crespy
*ae
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: crespy@mpip-mainz.mpg.de
bInstitute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dDepartment of Chemical Engineering, School of Environmental and Chemical Engineering, Shanghai University, Shangda Road 99, 200444, Shanghai, P. R. China
eVidyasirimedhi Institute of Science and Technology (VISTEC), 555 Moo 1 Payupnai, Wangchan, Rayong 21210, Thailand
First published on 26th April 2016
Abstract
Materials capable of controlling the release of functional agents are promising for drug delivery and corrosion protection. A dual-responsive multicompartment nanostructure is designed by embedding redox-responsive nanocapsules in pH-responsive nanofibers by colloid-electrospinning. This combination allows an enhanced control over the release of payloads directed by properties of nanocontainers and nanofibers.
Materials capable of sensing the changes of their surrounding environment, responding through conformational and/or chemical changes, and releasing functional agents to maintain or recover their functions are promising for drug delivery, self-healing, and corrosion protection.1 Stimuli-responsive nanocontainers are especially interesting for controlled release of functional agents encapsulated in their core. In the fields of biomedicine2 and corrosion protection,3 ideal nanocontainers shall be able to release payloads selectively upon specific conditions. Stimuli-responsive nanocarriers are playing an important role because they allow a controlled and specific delivery of payloads to the surrounding medium upon reception of an external signal.1
Although a cornucopia of methods for the preparation of nanocontainers is reported in the literature, few exist that allows an in situ encapsulation of payloads during the formation of nanocontainers with a liquid core. The miniemulsion process, in which stable nanodroplets are formed by high shearing forces,4 can fulfill these conditions. Thus, stimuli-responsive nanocapsules were successfully obtained in miniemulsion either by using a stimuli-responsive shell,5 a switchable core,6 or a combination of both.7,8 Functional payloads can either be physically entrapped in the core of nanocarriers,5,9 adsorbed on the nanocontainers surface,8 or chemical bound to the shell of the nanocontainers.7 Thereby a selective release of payloads can be realized, i.e. the release of one payload upon one stimulus from a pool of different payloads.7,8
Nanocontainers however are usually difficult to be separated from a surrounding medium. The colloid-electrospinning technique offers the possibility to immobilize nanoparticles in nanofibers,10 hence allowing the construction of a hierarchical structure. The fact that these materials can be easily separated could be advantageous for catalysis applications,11 while the double barrier offered by the material of nanofibers and the material of nanocontainers shell was found beneficial for the protection of dyes for photon upconversion.12
Nanoparticles were electrospun in thermoresponsive fibers for releasing payloads upon increase of temperature. The heat needed to collapse the fibers hydrogel composed of temperature-responsive polymer was generated indirectly thanks to the adsorption of near-infrared light by gold nanoparticles embedded in the fibers.13 An alternating magnetic field could be used as trigger for the payloads release when magnetite was used instead of gold nanoparticles.14 No combination of stimuli-response from both nanocontainers and nanofibers matrix was described.
We aim here at exploring the combination of stimuli-responsive properties of nanocapsules and nanofibers for controlling the release profiles of payloads encapsulated in the capsules. Redox-responsive silica nanocapsules (SiNCs) are incorporated in pH-responsive nanofibers by colloid-electrospinning. This combination of responsiveness is especially promising for corrosion protection because changes of pH15 and redox potential16 occur upon onset of corrosion. To verify the applicability of our nanostructured system for the controlled release of payloads, 2-mercaptobenzothiazole (MBT) was selected as functional payload because it is widely used as corrosion inhibitor.17
The SiNCs were synthesized by using miniemulsion droplets as templates for the hydrolysis and condensation of tetraethoxysilane (TEOS) and an alkoxysilane containing tetrasulfide bonds (TESPT) (see Fig. S1†). The encapsulated corrosion inhibitor MBT was dissolved in the organic liquid forming the droplets and is hence directly encapsulated in the core of the nanocontainers. The loading efficiency of MBT in silica nanocapsules was calculated to be 96% by using UV-vis spectroscopy. A hydrodynamic diameter of 127 ± 43 nm of the SiNCs was evaluated by dynamic light scattering. Well-defined core–shell morphologies of the capsules were identified by SEM and TEM as shown in Fig. S1.†
The SiNCs were then electrospun in the presence of aqueous polymer solutions to form materials composed of the nanocapsules embedded in hydrophilic nanofibers (see Fig. 1a). A strong electrical field was applied to the dispersion which was fed at a constant rate. Once the electrostatic force overcame the surface tension of the droplets, the dispersion was ejected towards the collector. The solvent was evaporated on the way to the collector and nanofibers were deposited on the collector as a non-woven.18 Three polyvinyl alcohol/polyacrylic acid (PVA/PAA) nanofiber samples with –COOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :–OH molar ratios of 0:1, 1:1 and 2:1 were prepared and named NF-0, NF-1 and NF-2, respectively. Uniform fibers with diameters ∼300 nm were obtained for all three samples (see Fig. 1e–g). The SiNCs (∼24 wt%) were distributed in the nanofibers as indicated by TEM (see Fig. 1b–d). The theoretical loading percentage of MBT in the fibers is 0.31 wt%. Polymer nanoparticles are usually difficult to be identified by electron microscopy when they are embedded in polymer fibers. Normally, fluorescence microscopy and fluorescently labeled nanoparticles are used to cope with this problem.12,19 Remarkably in our case, the core–shell structure of the nanocapsules can be still recognized after being embedded in the nanofibers (see Fig. 1b–d).
:–OH molar ratios of 0:1, 1:1 and 2:1 were prepared and named NF-0, NF-1 and NF-2, respectively. Uniform fibers with diameters ∼300 nm were obtained for all three samples (see Fig. 1e–g). The SiNCs (∼24 wt%) were distributed in the nanofibers as indicated by TEM (see Fig. 1b–d). The theoretical loading percentage of MBT in the fibers is 0.31 wt%. Polymer nanoparticles are usually difficult to be identified by electron microscopy when they are embedded in polymer fibers. Normally, fluorescence microscopy and fluorescently labeled nanoparticles are used to cope with this problem.12,19 Remarkably in our case, the core–shell structure of the nanocapsules can be still recognized after being embedded in the nanofibers (see Fig. 1b–d).
 | ||
| Fig. 1 (a) Schematic illustration of the preparation of multicompartment nanofibers; (b–d) TEM and (e–g) SEM micrographs of nanofibers with different ratio of PAA and PVA; (b, e): NF-0, (c, f): NF-1, and (d, g): NF-2. | ||
The responsivity of the produced materials should be given by the fact that the SiNCs and the fibers are redox- and pH-responsive, respectively (see Fig. 2a). Because the release studies were performed in water, the PVA fibers were crosslinked with glutaraldehyde. Compared with crosslinking by esterification that requires the sample to be heated,20 the PVA/PAA fibers were crosslinked at room temperature in our study. Moreover, the mixture of PVA and PAA yields also fibers with strong hydrogen bonds.21 This method hence ensures that temperature-sensitive payloads can be loaded in the fibers. pH- and redox-responsive properties of the nanofibers were investigated by soaking the fibers in media of different pH values or/and with different concentrations of the water-soluble reducing agent tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl), which is highly effective for the reduction of disulfide bonds.22 The release of MBT from the fibers was then determined by UV-Vis spectroscopy.
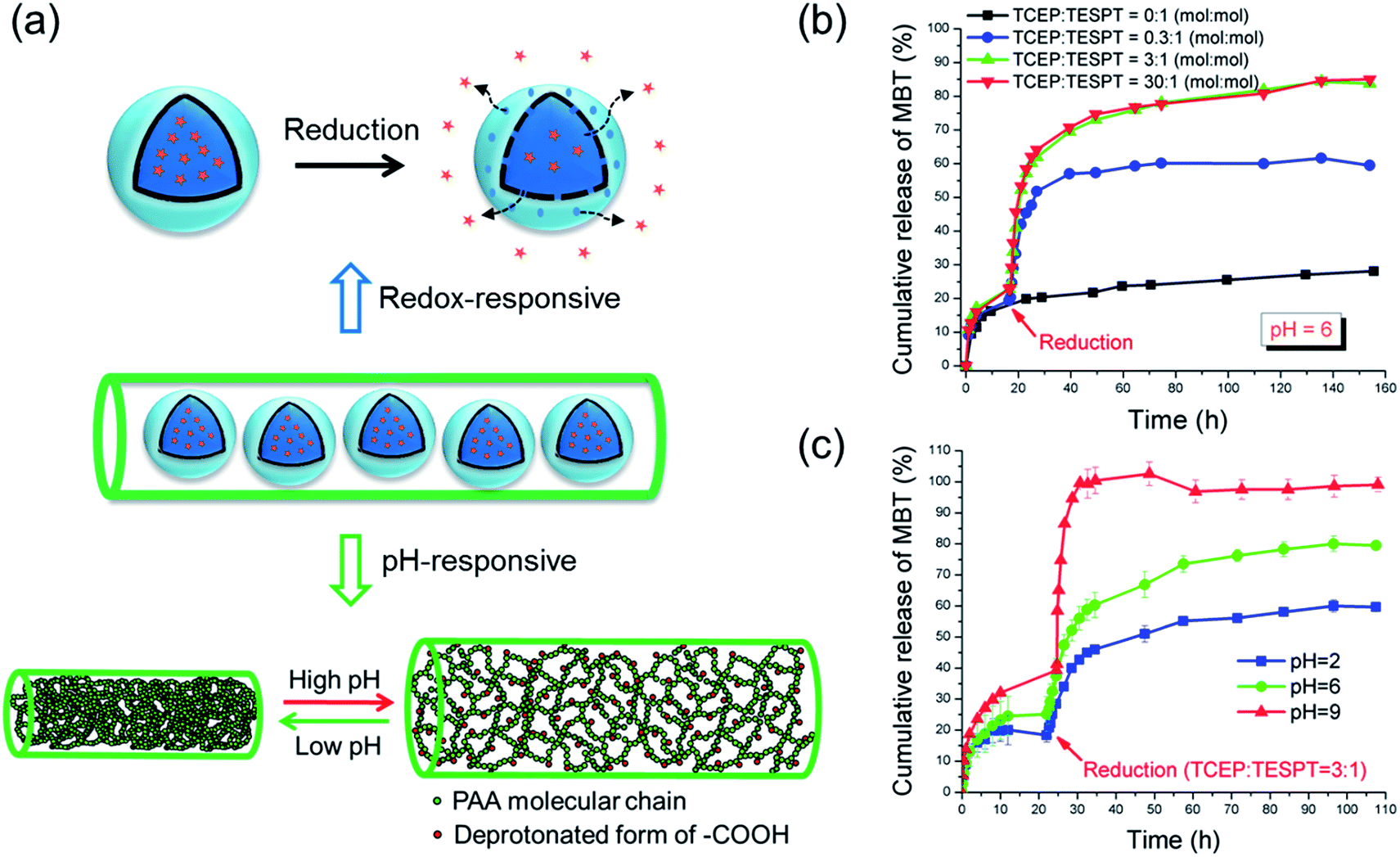 | ||
| Fig. 2 (a) Schematic illustration of mechanism of redox- and pH-responsive release from nanofibers; (b) Redox-responsive release of MBT from NF-1; (c) pH-responsive release of MBT from NF-1. | ||
When no reducing trigger was applied, the fibers NF-1 showed a non-specific release of ∼27% MBT (compared to the initial amount of MBT in the fibers) after 160 h (see Fig. 2b). This non-specific release, i.e. the release of MBT without reducing agent, was dramatically accelerated by adding 0.07 mmol L−1 TCEP. This acceleration was attributed to the reduction of tetrasulfide linkages by TCEP, which led to an increase of the shell permeability. When the concentration of TCEP was further increased, both release rate and release amount of MBT increased accordingly (see Fig. 2b). The TCEP concentration-dependent release behavior mainly depends on the diffusion rate of TCEP to the tetrasulfide bonds. When a higher concentration of TCEP is used, a higher gradient of concentration between the dialysis solution and the silica shell is created. Therefore a comparatively higher amount of TCEP can reach and reduce the tetrasulfide bonds in a shorter time. No distinguishable acceleration was observed when TCEP concentration was further increased from 0.7 to 7 mmol L−1, indicating that after a certain concentration the release was not dominated anymore by the diffusion of TCEP. Remarkably, the redox-responsive groups were still addressable although they were embedded in the nanofibers.
The pH-responsive release of MBT was studied at various pH values but with the same concentration of reducing agent (TCEP:TESPT = 3:1, see Fig. 2c). The nanofibers NF-1 were first immersed in buffer solutions to study the diffusion of MBT from fibers at different pH conditions. In this case, the release of MBT mainly depends on the swelling of the fibers. Indeed, the pH sensitivity of the PVA/PAA fibers is affected by the protonation state of the carboxylic groups of PAA, which is a weak acid with an intrinsic pKa of ∼4.7.23 Therefore, the fibers were swollen at higher pH and unswollen at low pH values. In the first ∼22 h, the diffusion of MBT molecules through the PVA/PAA fiber matrix was higher at high pH value as shown in Fig. 2c. As expected, an accelerated release of MBT and higher released amount at high pH was observed when the reducing agent was added. This facilitated release can be explained by the higher swelling degree of PAA/PVA nanofibers at high pH value that allows the TCEP to diffuse more easily in the nanofibers (see Fig. 2a).
To select a suitable reducing agent for a specific system is very important. During the study of the MBT release at various pH values, we found that the release can be significantly accelerated by adding the reducing agent TCEP at pH values of 2 and 6. However, when we increased the pH value to 9, no obvious increase of the released amount of MBT was observed when TCEP was added. This was attributed to the oxidation of TCEP at higher pH as reported previously in the literature.24 To solve this problem, the reducing agent DTT was used at pH 9 instead of TCEP. As expected, the release was accelerated when DTT was added (see Fig. S2†). The release profile of the MBT from the nanocontainers can be therefore controlled by the pH-responsive properties of the fibers and both the released amount and the release speed increase with increasing pH value of the surrounding medium.
To further study how the fibers matrix is influencing the release profile of payloads from embedded nanocontainers, the ratio between the PAA and PVA was varied. The release profile from PVA fibers, i.e. non-responsive fibers, is drawn for the sake of comparison with the release profiles of the PVA/PAA fibers (see Fig. 3).
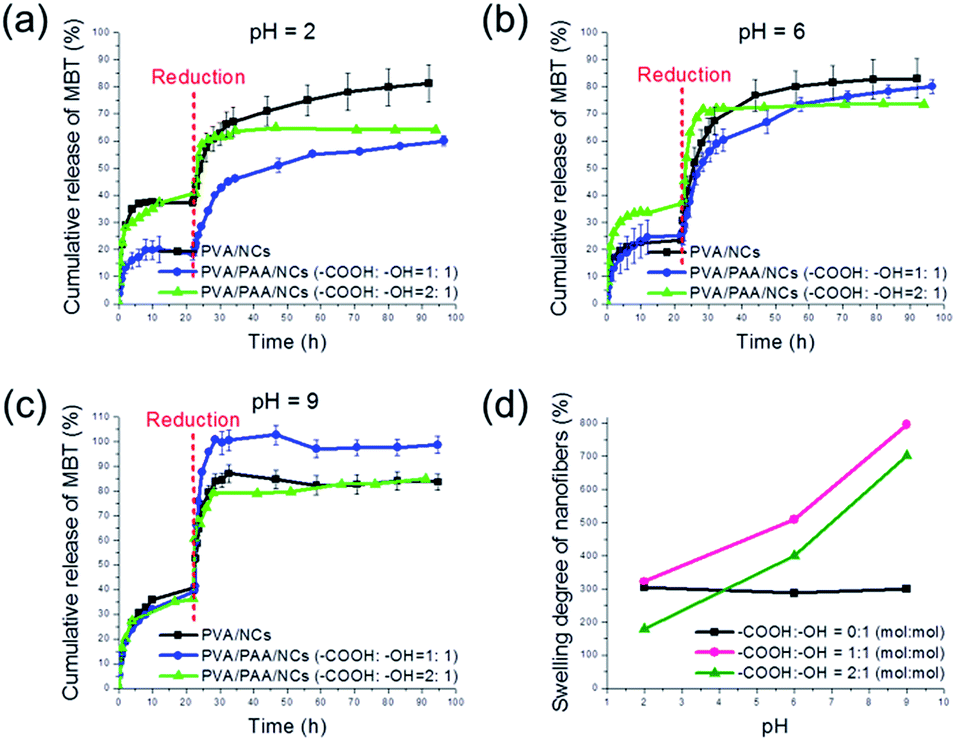 | ||
| Fig. 3 Release profiles of MBT from PVA/PAA nanofibers with different content of PAA at (a) pH = 2, (b) pH = 6 and (c) pH = 9. Reducing conditions at different pH were created with molar ratio of TCEP:TESPT = 3:1. (d) Swelling degree of PVA/PAA nanofibers with various PAA contents. | ||
For the PVA nanofibers (NF-0), ∼80% of loaded MBT was released at pH = 2, 6, and 9 at the equilibrium (see Fig. 3a–c). This similar equilibrium concentration in all three pH conditions is consistent with measurement of the swelling degree. Indeed, the fibers NF-0 exhibited a similar swelling degree of ∼300% at the different pH values used in this study (see Fig. 3d).
For the PVA/PAA nanofibers (NF-1), as mentioned above in Fig. 2c, a pH-dependent release behavior of MBT was observed. At pH = 2, the release of MBT was retarded compared with the pure PVA fibers. When the TCEP was added, an equilibrium concentration of ∼60% of MBT was observed compared with 80% of release from NF-0 (see Fig. 3a). The difference can be explained by the physical crosslinking occurring because of the hydrogen bonds between PVA and PAA. At pH = 6, fiber NF-1 shows a release of ∼80% in 98 h (see Fig. 3b). Remarkably, a fast and almost 100% release from NF-1 was observed at pH = 9 (see Fig. 3c). The facilitated release can be attributed to the higher swelling degree resulting from the deprotonation of the –COOH of PAA at high pH (see Fig. 3d). Indeed, the swelling degree of the nanofibers NF-1 was measured to be ∼500% at pH = 6 and ∼800% at pH = 9.
In the case of PVA/PAA nanofibers with the molar ratio –COOH:–OH = 2:1 (NF-2), an increased equilibrium concentration was also observed with increasing the pH of buffer compared to the non-responsive fibers NF-0 (see Fig. 3a–c). At pH = 2, the release of MBT was retarded, only an equilibrium concentration of ∼60% of MBT was obtained (see Fig. 3a). At pH = 6, it shows a similar behavior with both NF-0 and NF-1 (see Fig. 3b). However, the release at pH = 9 is ∼80% when reached equilibrium, which is less than the 100% release of NF-1 (see Fig. 3c). This lower equilibrium concentration is consistent with the swelling degree results. NF-2 shows a lower swelling degree than that of NF-1 at various pH values (see Fig. 3d). The fact that the fibers NF-2 are actually less responsive than NF-1 although they contain more PAA can be attributed to the higher physical crosslinking density in the fibers NF-2. The hydrogel nanofibers possess then a lower free volume and the diffusion of TCEP and MBT is less facilitated as for the fibers NF-1.
Conclusions
In summary, nanomaterials with a hierarchical structure composed of nanocapsules-in-nanofibers were fabricated by colloid-electrospinning. A corrosion inhibitor was encapsulated in redox-responsive nanocapsules prior to their embedding in the nanofibers. Release profiles of the corrosion inhibitor upon different reduction conditions indicate that the fibers matrix plays a major role on both the released amount and the release speed of the encapsulated payload. The use of a pH-responsive matrix was found beneficial to provide an additional stimuli-specific release behavior to the system so that it is sensitive to both the pH value and the concentration of reducing agent in the surrounding medium. This gives also the possibility to fabricate delivery systems that needs to be simultaneously activated by two different stimuli, thus preventing unnecessary leakage of the payload if only one stimulus is activated. The presented concept is general and allows for a flexible combination of properties for the nanocontainers and nanofibers. These advantages make the nanocapsules-in-nanofibers constructs promising candidates for controlled release of payloads for self-healing and drug delivery applications. Because both pH value and redox potential are changing on corrosion onset, the material could be applied for corrosion protection.Acknowledgements
We acknowledge the financial support from ‘‘MPG-CAS Joint Doctoral Promotion Program (DPP)’’.References
- M. A. C. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed.
- D. Shchukin and H. Möhwald, Science, 2013, 341, 1458 CrossRef CAS PubMed.
- D. Schaeffel, R. H. Staff, H.-J. Butt, K. Landfester, D. Crespy and K. Koynov, Nano Lett., 2012, 12, 6012 CrossRef CAS PubMed.
- L.-P. Lv, Y. Zhao, N. Vilbrandt, M. Gallei, A. Vimalanandan, M. Rohwerder, K. Landfester and D. Crespy, J. Am. Chem. Soc., 2013, 135, 14198 CrossRef CAS PubMed.
- Y. Zhao, K. Landfester and D. Crespy, Soft Matter, 2012, 8, 11687 RSC.
- L.-P. Lv, K. Landfester and D. Crespy, Chem. Mater., 2014, 26, 3351 CrossRef CAS.
- R. H. Staff, M. Gallei, K. Landfester and D. Crespy, Macromolecules, 2014, 47, 4876 CrossRef CAS.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song and A. G. Schwartz, Nat. Mater., 2009, 8, 935 CrossRef CAS PubMed.
- D. Crespy, K. Friedemann and A. M. Popa, Macromol. Rapid Commun., 2012, 33, 1978 CrossRef CAS PubMed.
- N. Horzum, M. Mari, M. Wagner, G. Fortunato, A.-M. Popa, M. M. Demir, K. Landfester, D. Crespy and R. Muñoz-Espí, RSC Adv., 2015, 5, 37340 RSC.
- C. Wohnhaas, K. Friedemann, D. Busko, K. Landfester, S. Baluschev, D. Crespy and A. Turshatov, ACS Macro Lett., 2013, 2, 446 CrossRef CAS.
- V. V. Ramanan, K. C. Hribar, J. S. Katz and J. A. Burdick, Nanotechnology, 2011, 22, 494009 CrossRef PubMed.
- Y. J. Kim, M. Ebara and T. Aoyagi, Adv. Funct. Mater., 2013, 23, 5753 CrossRef CAS.
- T. H. Tran, A. Vimalanandan, G. Genchev, J. Fickert, K. Landfester, D. Crespy and M. Rohwerder, Adv. Mater., 2015, 27, 3825 CrossRef CAS PubMed.
- A. Vimalanandan, L. P. Lv, T. H. Tran, K. Landfester, D. Crespy and M. Rohwerder, Adv. Mater., 2013, 25, 6980 CrossRef CAS PubMed.
- M. Ohsawa and W. Suëtaka, Corros. Sci., 1979, 19, 709 CrossRef CAS.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670 CrossRef CAS PubMed.
- E. Jo, S. Lee, K. T. Kim, Y. S. Won, H. S. Kim, E. C. Cho and U. Jeong, Adv. Mater., 2009, 21, 968 CrossRef CAS.
- X. Jin and Y. L. Hsieh, Polymer, 2005, 46, 5149 CrossRef CAS.
- S. Sinha-Ray, Y. Zhang, D. Placke, C. M. Megaridis and A. L. Yarin, Langmuir, 2010, 26, 10243 CrossRef CAS PubMed.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648 CrossRef CAS.
- H. Byun, B. Hong, S. Y. Nam, S. Y. Jung, J. W. Rhim, S. B. Lee and G. Y. Moon, Macromol. Res., 2008, 16, 189 CrossRef CAS.
- J. C. Han and G. Y. Han, Anal. Biochem., 1994, 220, 5 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional characterization data. See DOI: 10.1039/c6ra05687c |
| This journal is © The Royal Society of Chemistry 2016 |