DOI:
10.1039/C6RA07621A
(Paper)
RSC Adv., 2016,
6, 63037-63048
Synthesis, thermal characterization and local structure studies of Gd doped Th0.7U0.3O2 using X-ray absorption spectroscopy†
Received
23rd March 2016
, Accepted 26th June 2016
First published on 27th June 2016
Abstract
(Th0.7−xU0.3Gdx)O2+y microspheres where x = 0, 0.05, 0.10 and 0.15 were prepared by internal gelation method. Good quality translucent microspheres of size ∼1.70 to 2.0 mm were obtained when low metal ([M]) and high [hexamethylenetetramine + urea]/[M] (R) concentration was used. The solid solutions prepared from these microspheres were subjected to thermal expansion studies which showed a gradual decrease in thermal expansion coefficient values with increase in Gd ion concentration. Specific heat capacities of mixed oxides were measured from 300 to 850 K and showed that the temperature dependence of the heat capacities of Gd doped samples is similar to that of the un-doped sample. The local structure studies by EXAFS showed that Gd–O, Gd–Th and longer Gd–O paths are similar to Th–O, Th–Th and longer Th–O path lengths. EXAFS studies showed higher oxygen coordination in the case of the 10% Gd substituted sample compared to the other samples.
1. Introduction
India has vast deposits of thorium over uranium and thorium will play an important role in the future Indian nuclear energy program. Construction of thorium based 300 MWe Advanced Heavy Water Reactor (AHWR) is an important step for this purpose. It is proposed to use (Th, U)O2 or (Th, Pu)O2 containing 3–4% of uranium or plutonium dioxides as fuel materials for the advanced heavy water reactors (AHWRs).1,2 It has been reported that the mixed oxide comprising a mixture of 70 wt% ThO2 and 30 wt% UO2 (U is enriched to ∼19.5 wt% U-235) has several advantages including higher burnup over UO2 based fuel.3 Solid solutions of doped thorium uranium oxide are of importance for the nuclear industry. Burnable neutron poisons are used in nuclear reactors to produce a more level distribution of power in the reactor core and to reduce to necessity for a large control system. A neutron poison (also called a neutron absorber or a nuclear poison) is a substance with a large neutron absorption cross-section, in applications such as nuclear reactors. As gadolinium is an excellent burnable neutron poison, incorporation of gadolinium in nuclear power reactor fuel is recommended from the point of reactivity compensation and adjustment of power distribution enabling, longer fuel cycles and optimized fuel utilization in LWR.4,5 The thermal margin in LWR is increased by introducing thorium along with gadolinium–uranium oxide pellets.6 Thoria urania containing 5–10 wt% of Gd is being proposed as a burnable absorber fuel for LEU-AHWR. The most important application of the addition of dopants to these mixed oxides is the production of neutron-absorbing fuel (NAF), which is used to optimize the reactivity in nuclear power reactors by reducing the reactivity of fresh fuel assemblies at the beginning of their active life in the reactor.7–9 For the production of NAFs, gadolinia (Gd2O3) is highly suitable as a dopant due to the large neutron absorption cross section of the gadolinium isotopes 155Gd and 157Gd.
Several extensive studies pertaining to phase characteristics and thermal expansion of UO2–Gd2O3 system have been reported earlier workers.10–12 Thoria forms a complete range of solid solution with urania. But the solubility of trivalent lanthanides in ThO2 is limited to few mole%. It was reported that the solubility of lanthanides decreases with decrease in their ionic size and increases with rise in the temperature.13 They have also further shown that in ThO2, gadolinium dissolves up to 30 mol% at 1200 °C and the solubility increases up to 40% at 1400 °C. Recently, Meera et al.14 studied the crystal chemistry and thermal expansion of thoria–urania–gadolinia system in which they had restricted the concentration of uranium in the solid solution up to 7 mole%.
Extensive work has already been published in the past on the preparation of Gd2O3-doped UO2 and Gd2O3-doped ThO2. Conventionally, these oxides are prepared by powder metallurgical route.15–22 But powder metallurgical route involves radiotoxic dust generation and the pellets derived by this method lack micro homogeneity. Many workers had demonstrated a co-precipitation method for obtaining this kind of fuel that offers superior micro homogeneity.23–29 Due to the high radio toxicity and man rem problem associated with these kinds of oxides, sol–gel process offers many advantages, as this process is highly amenable for automation and remote handling. In the past, Gündüz et al.30,31 demonstrated sol–gel derived methods for the preparation of UO2–Gd2O3 fuel materials. We report preparation of (Th0.7−xU0.3Gdx)O2+y microspheres where x = 0, 0.05, 0.10 and 0.15 by internal gelation method32 for the first time. The thermo-physical properties of sintered pellets derived from these microspheres such as heat capacity and thermal expansion behavior is also studied.
It has been reported by earlier workers that when Gd3+ is incorporated in thoria–urania solid solution, U4+ is getting oxidized to U5+ and U6+ and also, the concentration of U6+ increases in the solid solution when Gd3+ ion concentration increases. They have come to this conclusion on the basis of oxygen to metal atomic ratio O/M14 they have observed in the samples. There are many methods available for monitoring the dopant ion and their effect in the crystal lattice. X-ray absorption spectroscopy (XAS) is particularly and routinely employed for the determination of the dopant location in the crystal lattice. Moreover, it also has the advantage of determining the coordination sites composed of elements possessing nearly equal X-ray scattering power. The local structures were probed using X-ray absorption spectroscopy.
2. Experimental details
2.1. Preparation of (Th0.7−xU0.3Gdx)O2+y microspheres
The flow-sheet used for the preparation of thoria–urania–gadolinia microspheres in the present study is shown in Fig. 1. The principal steps involved are:
 |
| | Fig. 1 Flow-sheet for the preparation of (Th0.7−xU0.3Gdx)O2+y pellets. | |
– Preparation of feed solution.
– Conversion of feed droplets into solid gel spheres (gelation).
– Washing of gel spheres, heat treatment of the gel spheres to obtain calcined Th0.7−xU0.3GdxO2+y microspheres.
The feed solution was prepared by mixing pre-cooled metal nitrate solutions of thorium (3 M) (≥98%; supplied by Indian Rare earth Ltd.), uranium (3 M) (Nuclear grade; supplied by Atomic Fuels Division, BARC) and gadolinium (2.6 M) (≥99.0%; supplied by Indian Rare earth Ltd.) at required proportions with pre-cooled A.R. grade hexamethylenetetramine (HMTA)–urea (≥99% supplied by Sigma Aldrich) solution. The total metal ion concentration [Th + U + Gd] ([M]) in the feed solution was varied from 1.0 to 1.30 M. For each metal ion concentration the [HMTA, urea]/[M] ratio (R) was varied in a range from 1.20 to 1.60. The feed solution was forced through a capillary of ∼1 mm diameter. The droplets converted into gel particles when contacted with hot silicone oil, circulated in a glass column at 90 °C. A three feet gelation column was used for carrying out the gelation which could provide residence time of 15–18 s for gelation to occur when the droplets are dispersed. The gel spheres were washed with CCl4 followed by 3 M NH4OH and dried at 100 °C for 6 h followed by 250 °C for 4 h. These were then calcined at 500 °C in air at a heating rate of 5 °C min. The pellets derived from these microspheres were sintered in Ar + 8% H2 at 1400 °C for 2 h. Alternatively, one set of pellets were sintered in air and another in vacuum (10−5 mbar) at 1400 °C for 2 h. The sintering was carried out at a heating rate of 5 °C min in all sintering atmospheres. X-ray diffraction (XRD) measurements were carried out by Rigaku MINIFLEX 600 X-ray diffractometer (h–2h geometry) using Cu Ka radiation (k = 1.5406 Å) with the scan rate of 1° min−1 for phase identification. The densities of the green as well as sintered pellets were determined by geometrical method. SEM (Philips make, Model: XL-30) investigations were carried out, on these samples to study the microstructure. For microstructural examination, a thin layer of gold (∼100 Å) was coated on the sintered pellets by thermal evaporation in a vacuum coating unit. A vacuum of 3.5 × 10−5 mbar was maintained using a diffusion pump having a rotary backing. A measured quantity of gold wire (99.99% pure) was wrapped on a tungsten wire which in turn was heated to evaporate the gold wire.
2.2. Analysis of oxygen to metal atomic ratio (O/U) and oxygen non stoichiometry (y)
O/U of all samples heated in air and reduced in Ar + 8% H2 were determined using chemical analysis. For this, known quantities of the oxide was dissolved in conc. phosphoric acid and total uranium as well as U(IV) in these solutions were analyzed by potentiometric titration using modified Davies and Gray method.33 Assuming that the Th and Gd in the solid solution is present only in +4 and +3 state respectively, and oxygen in −2 state, the O/U ratio was determined as follows.
| O/U = (3nU(VI) + 2nU(IV))/(nU(VI) + nU(IV)) |
where nU(VI) and nU(IV) are number of moles of U(VI) and U(IV), respectively. The O/M of Th0.7−xU0.3GdxO2+y was calculated as follows
| O/M = (0.7 − x) × 2 + 0.3 × O/U + 1.5 × x |
2.3. Evaluation of thermo-physical properties
The reducibility of air calcined microspheres was studied by a TPDRO-1100 instrument (Thermo Quest, Italy). Temperature Programmed Reduction (TPR) profiles was recorded by heating the thoria–urania–gadolinia microspheres up to 1000 °C in a heating rate of 10 °C min. Ar–8% H2 and high purity Ar was flown at reactive side and carrier side respectively in a flow rate of 10 mL min−1.
To study the thermal expansion behavior, high temperature X-ray diffraction (HTXRD) patterns of sintered samples were recorded using HDK-2.4 Buhler high temperature attachment to a Stoe theta–theta X-ray diffractometer using monochromatised Cu Kα1 radiation (λ = 1.5406 Å). The furnace temperature was measured by Pt/Pt–13% Rh thermocouple and was controlled by a PID-type temperature controller within ±1 K during the measurements. The samples were equilibrated for 15 min at each temperature before the measurement of X-ray data. The powder sample was mounted on a tantalum strip that was resistively heated at a programmed rate. Coefficients of axial and volume thermal expansions were measured in vacuum of about 10−5 mbar in the temperature range from 298 to 1273 K in steps of 100 K. Further details on the instrumentation about HTXRD used in the present study are given elsewhere.34 The lattice parameters were refined using least square method using a computer program35 and were obtained within the precision of ±0.001 Å. Silicone powder was used at room temperature for calibration prior to the measurements of the lattice parameters and the thermal expansion of MgO was determined by the same method as mentioned above. The results obtained were within 5% of the reported values.36
Specific heat capacities of the above samples were determined by the classical three step method in continuous heating mode using heat flux type Differential Scanning Calorimeter using (model DSC 1/700) of M/S Mettler Toledo GmbH, Switzerland from temperature range 300 to 873 K at a heating rate of 10 K min−1. These experiments were carried out in high purity Ar gas atmosphere using a flow rate of 20 mL min−1. The sample and reference material (NIST synthetic sapphire (SRM 720)) were taken in two flat bottom Pt crucibles (vol. 70 μL) of identical masses with covering lids having small hole at the centre. About 150–200 mg sample was used for each experiment. The temperature, heat and τ lag calibration were carried out prior to the heat capacity measurements. An identical three-segment heating program was used for blank, sapphire and sample runs. Temperature and the heat flow calibrations were performed using In, Zn and Pb as standards.
2.4. Local structure studies of Th0.7−xU0.3GdxO2+y using X-ray absorption spectroscopy
Extended X-ray Absorption Fine Structure (EXAFS) measurements on Th0.7−xU0.3GdxO2+y samples where x = 0, 0.05, 0.10 and 0.15 were performed using Energy-Scanning EXAFS beam line (BL-9) at the INDUS-2 Synchrotron Source (2.5 GeV, 100 mA) at Raja Ramanna Centre for Advanced Technology (RRCAT), Indore, India37,38 to probe the local structure. This beam line operates in energy range of 4 KeV to 25 KeV. The beam line optics consist of a Rh/Pt coated collimating meridional cylindrical mirror and the collimated beam reflected by the mirror is monochromatized by a Si(111) (2d = 6.2709) based double crystal monochromator (DCM). The second crystal of the DCM is a sagittal cylinder with radius of curvature in the range 1.28–12.91 meters which provides horizontal focusing to the beam while another Rh/Pt coated bendable post mirror facing down is used for vertical focusing of the beam at the sample position. Rejection of the higher harmonics content in the X-ray beam is performed by detuning the second crystal of DCM.
For the EXAFS studies, appropriate weight of samples were taken in powder form to obtain a reasonable edge jump and were mixed thoroughly with cellulose powder to obtain total weight of 100 mg and homogenous pellets of 15 mm diameter pellets were made.
3. Results and discussion
3.1. Preparation of thoria–urania–gadolinia microspheres
For the preparation of thoria–urania–gadolinia microspheres, we have used a glass column of dimension 3 feet length and diameter 3 inches approximately. This feed solution was then converted into droplets by forcing the solution through a 1 mm ID SS capillary. It was observed that most of the droplets were not gelling within the length of the gelation column or partially gelling into a soft mass. The gelation could be obtained in the above mentioned column by mixing the metal nitrate solutions and HMTA, urea solution and keeping the mixed broth for different duration of time. This time delay before dispersion of the broth was responsible for partial hydrolysis of the metal, enabling gelation within the length of the column. The wet gel microspheres were light yellowish in colour with the texture varying from translucent to opaque. The wet gel microspheres were of dimension 1.70–2.0 mm in diameter. Fig. 2 shows photographs of gel microspheres containing varying amounts of Gd. The translucent microspheres were hard, but the opaque ones were softer.
 |
| | Fig. 2 Gel microspheres prepared by internal gelation method (a) thoria urania (b) thoria urania containing 5% of Gd (c) thoria urania containing 10% of Gd and (d) thoria urania containing 15% of Gd. | |
The variation of physical characteristics of the microspheres as a function of feed composition containing 15% of Gd is tabulated as shown in Table 1. For a given metal ion concentration, the formed gel changed from soft opaque to hard translucent appearance with the increase in molar ratio R. It can be seen that for [M] = 1.0 & 1.10 with high R values gave translucent microspheres (Table 1). Whereas, the opaque compositions were obtained with moderate R values. A similar trend in the gelation behavior was observed for compositions containing 5 and 10% of Gd concentrations. For each of the metal ion concentrations from 1.0 to 1.30 M, spherical gel particles were obtained. Pai et al.32 reported that the nature of the gel varied from soft opaque gel particles to hard opaque ones and at last to hard translucent particles, when R was increased in their study to prepare ThO2 microspheres containing 3 mole% of uranium. In the present study, the gel spheres obtained were translucent when higher R values were used. The lower R values and higher metal concentration could not yield opaque microspheres as the gelation was not complete for these compositions. Lloyd et al.39 studied gelation aspects for urania and attributed this effect to crystal habit and crystallization behaviour of urania. They reported that with the increase in R for a given [U], the microspheres progressively became smaller, less opaque and darker in colour suggesting that the crystallite size of the precipitated urania decreases as R increases. A probable explanation for similar behaviour in the case of thoria–urania–gadolinia system could be given in the present investigation on the basis of kinetics of the hydrolysis reaction. The rate of reaction for a given concentration of metal ion, increases with the increase in concentration of gelation agent i.e.; HMTA + urea. This rate of reaction governs the size of crystallites formed in the gel. The high reaction rate caused by high R restricts the extent of growth of crystallites yielding hard translucent gel microspheres. On the other hand, the low reaction rate from low R yields soft opaque gel particles.
Table 1 Characteristics of (Th0.55U0.3Gd0.15)O2+y gel microspheresa
| Sr. No. |
[M] |
R |
Nature of gel obtained |
| [M] = Total metal ion concentration in mol L−1; R = [HMTA, urea]/[M]. |
| 1 |
1.0 |
1.3 |
No gel |
| 2 |
1.0 |
1.4 |
Soft opaque gel |
| 3 |
1.0 |
1.5 |
Translucent |
| 4 |
1.0 |
1.6 |
Translucent |
| 5 |
1.1 |
1.3 |
Soft opaque gel |
| 5 |
1.1 |
1.4 |
Soft opaque gel |
| 6 |
1.1 |
1.5 |
Translucent |
| 7 |
1.1 |
1.6 |
Translucent |
| 8 |
1.2 |
1.3 |
No gel |
| 9 |
1.2 |
1.4 |
Soft opaque gel |
| 10 |
1.3 |
1.3 |
No gel |
When Gd incorporates into the lattice of thoria–urania, the lattice contracts. The lattice contraction due to doping of Gd in ThO2 and UO2 forming (Th1−yGdy)O2−y/2 and (U1−yGdy)O2−x has been reported by several authors.10,25,29,40,41 Fig. 3(a) shows the room temperature XRD pattern of (Th0.7−xU0.3Gdx)O2+y pellet samples sintered in air, vacuum and Ar + 8% H2 atmosphere.
 |
| | Fig. 3 (a) Room temperature XRD pattern of (Th0.7−xU0.3Gdx)O2+y oxides. (b) Room temperature XRD pattern of (Th0.7−xU0.3Gdx)O2+y oxides enlarged. | |
The room temperature X-ray diffraction patterns of (Th0.65U0.3Gd0.05)O2+y (TUG1), (Th0.60U0.3Gd010)O2+y (TUG2), and (Th0.55U0.3Gd0.15)O2+y (TUG3) are similar to (Th0.7U0.3)O2 (TU), except a shift in 2θ positions to marginally higher angles. This shift in the peak positions to higher 2θ values with increase in Gd content could be clearly seen from Fig. 3(b) which is the enlarged version of Fig. 3(a). All the samples sintered in air, vacuum and Ar + 8% H2 atmosphere were crystallized in to cubic fluorite lattice (Face Centred Cubic; CaF2 type). As reported for Gd doping in ThO2 and UO2, the lattice parameter decreases with increase in Gd content in all the samples under all sintering conditions. When Gd3+ is substituted for Th4+ ion in the cubic-fluorite (F) lattice, the oxygen deficiency is created in oxygen sub lattice to form defect fluorite lattice. Even though the size of Gd3+ is slightly larger than Th4+ (for 8 co-ordination the ionic radii of Th4+ = 1.05 Å and Gd3+ = 1.052 Å), the lattice parameter found to decrease from 5.5477 Å for (Th0.7U0.3)O2 to 5.5109 Å for 15% Gd substituted sample in air atmosphere. A similar linear decrease could be obtained in the lattice parameters for the sample sintered in vacuum and Ar + 8% H2. Fig. 4 shows a plot of lattice parameters of TU, TUG1, TUG2 and TUG3 with Gd content obtained on heating in air, vacuum and Ar + 8% H2 atmospheres. From the plot we can see that for a given concentration of Gd the lattice parameter of air sintered samples is the lowest and sample sintered in Ar + 8% H2 showed the highest lattice parameter. Also the lattice parameter of samples decreased with increase in Gd content for all sintering conditions. The thoria urania gadolinia solid solution is formed by random distribution of cations in the cation sub lattice. The decrease in the lattice parameter as we increase the Gd concentration is can be explained as follows. When Th4+ is substituted with Gd3+ in the cation lattice some of the U4+ is oxidized to either U5+ or U6+ in order to maintain electrical neutrality. The ionic radius of U5+ with eight fold co-ordination is 0.088 nm (ref. 42) and that of U6+ is 0.086 nm.43 Therefore, the increase in lattice constant expected by doping of cation with larger ionic radius is overwhelmed by decrease in the average ionic radius of uranium ions due to oxidation, assuming a random distribution of the cations in the cation sub lattice. Therefore, a net negative change in the lattice constant on increase in dopant concentration is observed.
 |
| | Fig. 4 Variation of lattice parameters of TU, TUG1, TUG2 and TUG3 with Gd content obtained on heating in air, vacuum and Ar + 8% H2 atmospheres. | |
3.2. O/M measurements and uranium valance estimation
Perfect stoichiometric oxides like ThO2, UO2 and Gd2O3 has O/M ratio 2.0, 2.0 and 1.5, respectively. But when a solid solution of these oxides is heated in different oxygen potentials, the oxygen non stoichiometry (y) arises due to the presence of uranium in the sample as uranium possesses multiple oxidation states whereas and Th & Gd possesses fixed valance of +4 and +3 respectively. Different non stoichiometry (y) was obtained by heating the samples in different gaseous environment having different oxygen potential such as air, vacuum and Ar + 8% H2. The O/M of the solid solutions were obtained from the chemical analysis of U(IV) and total uranium by potentiometry. U(VI)/U(total) ratio gives O/U, from which O/M is calculated using the formulae as discussed in Section 2.2 assuming oxidation states of Th4+ and Gd3+ are unchanged during heating conditions used in the present study. Table 2 shows the variation of lattice parameter, O/U, O/M and average uranium valance of these samples with varying amounts of Gd obtained by sintering in three different atmospheres, air, vacuum and Ar + 8% H2. Even though the uranium concentration in all the samples remained constant, different ‘y’ values were obtained due to the change in the Th/Gd ratio in the samples and change in the oxygen potential when sintered in different atmospheres. The chemical analysis carried out for determining the O/U in these samples show that the average valance of uranium in all the samples are more than 4.
Table 2 Lattice parameter, O/U, O/M and average uranium valence of Th0.7−xGdxU0.3O2+y samples where x = 0, 0.05, 0.10 and 0.15 obtained on heating at 1673 K in different atmospheric conditionsa
| Sample |
Lattice parameter (Å) |
Oxygen to uranium atomic ratio (O/U) |
Oxygen to metal ratio (O/M) |
Average uranium valance (Z) Z = 2 × O/U |
| Air |
Vacuum |
Ar + 8% H2 |
Air |
Vacuum |
Ar + 8% H2 |
Air |
Vacuum |
Ar + 8% H2 |
Air |
Vacuum |
Ar + 8% H2 |
| O/U = (3nU(VI) + 2nU(IV))/(nU(VI) + nU(IV)), where nU(VI) and nU(IV) are number of moles of U(VI) and U(IV), respectively. O/M = (0.7 − x) × 2 + 0.3 × O/U + 1.5 × x. |
| TU |
5.5477 |
5.5482 |
5.5551 |
2.596 |
2.142 |
2.030 |
2.179 |
2.042 |
2.009 |
5.192 |
4.284 |
4.060 |
| TUG1 |
5.5331 |
5.5360 |
5.5413 |
2.578 |
2.108 |
2.106 |
2.148 |
2.007 |
2.007 |
5.156 |
4.216 |
4.212 |
| TUG2 |
5.5221 |
5.5282 |
5.5284 |
2.578 |
2.327 |
2.313 |
2.123 |
2.048 |
2.044 |
5.156 |
4.654 |
4.626 |
| TUG3 |
5.5109 |
5.5143 |
5.5196 |
2.597 |
2.183 |
2.174 |
2.104 |
1.980 |
1.977 |
5.194 |
4.366 |
4.348 |
Durazzo et al.28 and Venkat Krishnan et al.44 have reported that the stoichiometry in (U, Gd)O2−x, synthesized in Ar–H2 atmosphere remains close to 2 up to 40 mol% GdO1.5 even though the expected stoichiometry was lesser than 2. They have concluded this mainly because of oxidation of U4+ to higher valence state which increased O/M of the ternary oxide. Meera Keskar et al.14 in their studies observed that O/M values for [(U0.1Th0.9)1−yGdy]O2+x are smaller than that of the values reported for (U, Gd)O2−x. The average valance of uranium observed in their samples containing 10 at% of Gd was close to 4. They have concluded this due to the lower concentration of uranium in oxides synthesized by them, which contribute to a lesser extent in increasing the O/M values. Whereas, with 30 at% uranium in our samples the uranium valance in our samples was more than 4. Even though the samples are reduced in Ar + 8% H2, because of the presence of Gd in the lattice, some of the uranium is getting oxidized into higher valence states once the samples reach to room temperature after sintering. It can be seen from the Table 2 that the O/U and O/M in the samples of having similar compositions are comparable when sintered in Ar + 8% H2 and vacuum atmospheres. Even though, oxidation of uranium takes place in these samples for charge balance, it is lesser compared to the air sintered samples. The O/M of sample sintered in these atmospheres is close to 2 where as the O/M of air sintered samples are more than 2.1 due to higher oxygen potential in this sintering condition.
3.3. Thermo-physical properties of thoria–urania–gadolinia
3.3.1. Thermal expansion studies. The thermal expansion study of TU, TUG1, TUG2 and TUG3 was carried out using HTXRD technique and coefficients of axial and volume thermal expansions were measured in vacuum from 298 to 1273 K. Table 3 lists the average thermal expansion coefficients measured of these oxides. It was observed that the values of average axial as well as volume thermal expansion coefficients of (Th0.7−xU0.3Gdx)O2+y oxides decreases with increase in Gd concentration. Mathews et al.22,45 reported a similar trend in thermal expansion coefficients using dilatometry and HTXRD.
Table 3 Lattice parameter and average thermal expansion coefficients of (Th0.7−xU0.3Gdx)O2+y
| Sample |
Crystal data |
Average thermal expansion coefficient ×10−6 (K−1) |
| a (Å) |
Vol. (Å)3 |
Axial |
Vol. |
| TU |
5.5594 |
171.8 |
9.415 |
28.82 |
| TUG1 |
5.5385 |
169.9 |
9.367 |
28.70 |
| TUG2 |
5.5300 |
169.1 |
9.147 |
28.34 |
| TUG3 |
5.5218 |
168.4 |
8.89 |
28.22 |
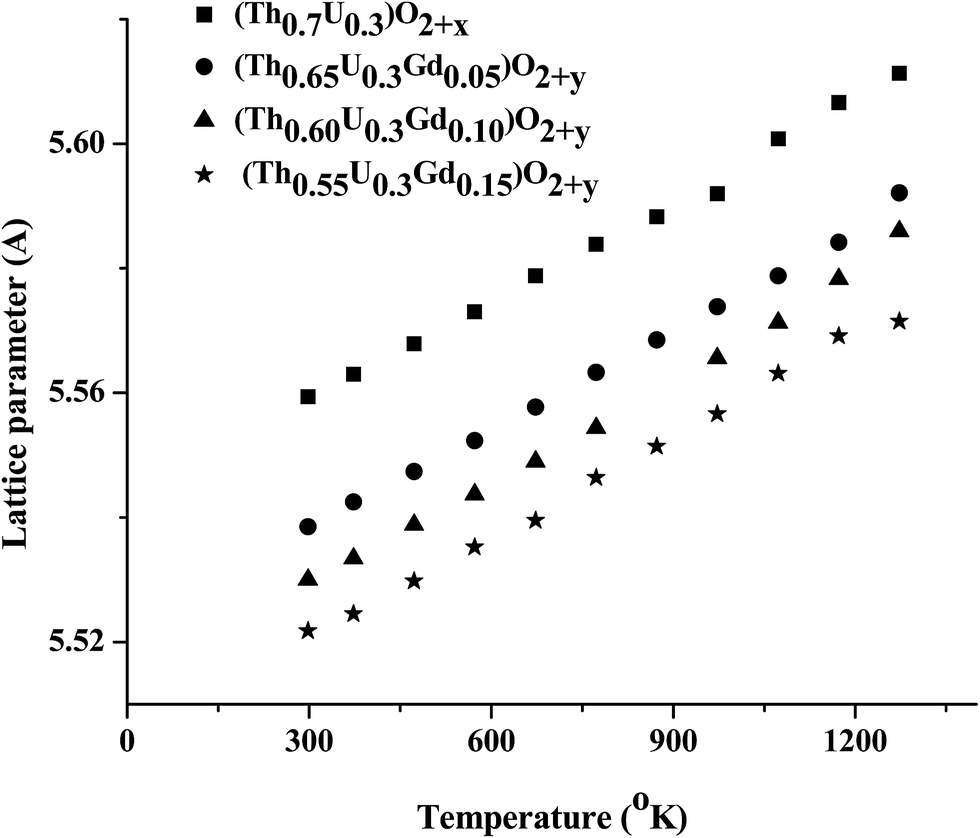
The axial (αa) and volume thermal expansion coefficient (αv) were derived from the equations αa = (ΔaT/a298) × (1/ΔT) and αv = (ΔvT/v298) × (1/ΔT), respectively. ΔaT is the difference between lattice parameter at temperature T and lattice parameter at room temperature. Similarly, ΔvT is the difference between cell volume at temperature T (a298) and cell volume at room temperature (v298) and ΔT is the corresponding temperature difference. The average thermal expansion coefficients of TU, TUG1, TUG2 and TUG3 showed decreasing trend with increase in Gd content which can be seen from Table 3. A similar trend was observed by Meera Keskar et al.14 in thermal expansion studies of thoria urania gadolinia system nevertheless, they have studied a lower concentration of uranium in their samples whereas, the uranium concentration in our samples were fixed at 30 at%. They have corroborated the decrease in thermal expansion with increase in Gd content due to (i) Body Centred Cubic (BCC) phase observed in their samples (in higher Gd content) and due to (ii) introduction of oxygen vacancy in the oxide when Gd is introduced in the lattice. Mathews et al.45 also reported that the BCC phase of Gd2O3 has lower thermal expansion values compared to fluorite phase of ThO2 up to 1673 K. The values of average thermal expansion coefficient values observed in our samples were higher compared to the results obtained by Meera Keskar et al. This may be due to the higher content of Gd present in their samples. Even though we did not observe any BCC phase in our samples due to Gd2O3 as we increased the Gd concentration, there observed a decrease in the thermal expansion coefficient value. This is because Gd is present as Gd3+ in the solid solution and as we increase the Gd content, defect concentration (oxygen vacancy) also increases. This might have decreased the thermal expansion in the doped samples. Room temperature XRD patterns of all the samples recorded before and after heating in vacuum at 1273 K are identical indicating no change in O/M. HTXRD patterns recorded from ambient temperature to 1273 K remained similar except for the shift in the X-ray line positions to lower 2θ values, indicating the expansion of lattice with rise in temperature. The lattice parameter of all compositions plotted against temperature is given in Fig. 5.
 |
| | Fig. 5 Variation of lattice parameters with temperature. | |
The variation in lattice parameter ‘a’ and volume ‘v’ of the oxides in the above graph were fitted to a second order polynomial expression,
where,
T denotes the absolute temperature in K.
P1,
P2,
P3 and
Q1,
Q2,
Q3 are the coefficients of the fitted
eqn (1) and
(2). These coefficients in the studied temperature range are presented in ESI Table 1.
†
3.3.2. Temperature programmed reduction studies. For studying the reducibility of the samples, the air-sintered samples were subjected to TPR (Fig. 6). The main feature of TPR is its capability of continuously monitoring the consecutive reaction of reducible species with increasing temperature after adsorbing the adsorbate gas on the surface.46–49 This study was intended to understand the species with different oxidation states present in the thoria–urania–gadolinia system. It will also give information about the nature of different reducible species. For this, the sample was placed in a quartz reactor system. Before start of the actual analysis the sample was pretreated under helium flow to 350 °C for 2 h. The reduction profile was thereafter recorded by heating the sample at a fixed rate (7 °C min−1) under the controlled flow of reactive gas mixture, i.e. 5% H2 in Ar up to 1000 °C. A thermal conductivity detector was employed to monitor the change in composition of reactive gas mixture with time. The sample was cooled after completion of the reduction. The reduction profiles of these samples showed only one reduction step corresponding to the reduction of UO2+x to UO2.0 present in these samples. It is reported in the literature that presence of rare earths in the urania lattice hinders the complete reduction of uranium to +4.50 This type of behavior was observed in the reduction profiles of our samples. Although, the uranium in all the samples was kept constant, the area under the peak, which signifies the amount of H2 consumed for reduction, was different indicating less O/M decrease. It was also observed that by doping Gd in Th site, the reduction profiles shifts to lower temperature. The reduction peak observed for thoria–urania sample was shifted down from 640 °C by 25–40 °C for Gd doped samples which showed that uranium could be reduced at lower temperature compared to samples which does not contain Gd.
 |
| | Fig. 6 TPR profiles of (Th0.7−xU0.3Gdx)O2+y oxides. | |
3.3.3. Heat capacity measurements. The molar heat capacities of (Th0.7−xU0.3Gdx)O2+y oxides were measured from 300 to 873 K using a differential scanning calorimeter. The specific heat capacity values for all the samples were plotted as a function of temperature and given in Fig. 7. It was observed that the temperature dependence of the heat capacities of Gd doped samples is similar to that of un-doped sample. The heat flow of thoria–urania–gadolinia sample doped with 10% Gd showed highest Cp values throughout the temperature range studied. The Cp values of 5 and 15 wt% Gd doped samples were lesser than that of thoria–urania. The individual values of heat capacities of different compositions were fitted by the least square method as a function of temperature in the form of polynomial expression Cp = A + BT + C/T2 and the values of the coefficients A, B and C are given in Table 4.
 |
| | Fig. 7 Molar heat capacity of (Th0.7−xU0.3Gdx)O2+y oxides. | |
Table 4 Coefficients of equation of heat capacities of (Th0.7−xU0.3Gdx)O2+y
| Sample |
Cpa (J K−1 mol−1) |
| A |
B |
C |
| Cp = A + BT + C/T2; where T is temperature in K. |
| TU |
67.8222 |
0.022 |
−8724 |
| TUG1 |
67.0689 |
0.018 |
−8393 |
| TUG2 |
72.4150 |
0.0165 |
−9311 |
| TUGd3 |
68.1258 |
0.0139 |
−10513 |
3.3.4. Calcination and sintering. The thoria–urania–gadolinia microspheres were calcined at 500 °C in air and pelletized into pellets of 10 mm dia and ∼8 mm height. Good quality high density pellets could be prepared from these calcined microspheres. These pellets were sintered at 1400 °C in Ar + 8% H2 atmosphere for 2 h. Table 5 shows the density of the green pellets and sintered pellets determined geometrically using a Screw gauge. It is assumed that the solid solution of gadolinium oxide in thorium–uranium oxide solid solution converts one uranium atom to +5 for charge compensation for each substituted dopant atom (gadolinium remains in its valence state +3). The metal fraction (x) of gadolinium was derived from the as-fabricated (added) content of Gd2O3. The T.D. of the specimens was calculated considering the composition of uranium and thorium measured by wet chemical method, the lattice parameter (a) as determined by the XRD measurements and taking the oxygen to metal ratio (O/M ratio) = 2.0. The atomic masses are taken as MTh = 232.038 g mol−1, MU = 238.028 g mol−1, MGd = 157.25 g mol−1 and MO = 15.999 g mol−1. Avogadro's number is taken as NA = 6.022 × 1023. The theoretical density of the sample was calculated according to the formula,| | |
Td = 4[(0.7 − x)MTh + xMGd + 0.3MU + 2MO]/(NAa3)
| (3) |
Table 5 Green density and sintered density of (Th0.7−xU0.3Gdx)O2+y pellets sintered at 1400 °C in Ar + 8% H2a
| Sample |
Green density (g cm−3) |
Observed sintered density (g cm−3) |
Theoretical density (T.D.) |
% T.D. |
| Td = 4[(0.7 − x)MTh + xMGd + 0.3MU + 2MO]/(NAa3), MTh = 232.038 g mol−1; MU = 238.028 g mol−1; MGd = 157.25 g mol−1; and MO = 15.999 g mol−1. Avogadro's number is taken as NA = 6.022 × 1023. |
| TU |
4.57 |
9.19 |
10.30 |
89.2 |
| TUG1 |
4.45 |
9.31 |
10.23 |
91.0 |
| TUG2 |
4.57 |
9.46 |
10.15 |
93.2 |
| TUG3 |
4.50 |
9.07 |
10.05 |
90.2 |
The density of pure thoria–urania samples is found to be less than 90% of T.D. when sintered in Ar + 8% H2 atmosphere at 1400 °C for 2 h. When the observed sintered densities of Gd substituted sample were compared with that of pure thoria urania, it can be seen that the sintered density first increases with Gd content up to 10% then decreases for 15% Gd substituted sample. The sintered pellets were examined under a Scanning Electron Microscope (SEM) for observing the microstructure. Fig. 8 shows the SEM photograph obtained by sintering these pellets in Ar + 8% H2 at 1400 °C for 2 h. An inhomogeneous microstructure with grain sizes ranging from 3–5 μm could be seen from the micrographs for samples containing 0 and 15% Gd whereas, the grain sizes of 5 and 10% Gd containing samples showed larger grains of 5–10 μm. The density of these sintered pellets determined by geometrical method corroborates the microstructure observed. When Th4+ is substituted by Gd3+ oxygen vacancy is being introduced. Due to this defect concentration, the sintering and grain growth occurs first with increase in Gd content up to 10% of Gd. The larger grain and higher density in 5 and 10% Gd containing samples may be attributed to this effect. The sintered density further decreases and the grain growth is further retarded as Gd increases to 15% in the sample as here the proportion of Gd3+ is more. Even though, uranium oxidation occurs here also, the proportion of oxygen interstitials due to the oxidation of uranium is offset by the oxygen vacancy due to substitution of more Gd3+ in place of Th4+.
 |
| | Fig. 8 Microstructure of sintered pellet of (a) TU; (b) TUG1; (c) TUG2 and (d) TUG3 in air at 1400 °C in Ar + 8% H2 for 2 h. | |
3.3.5. Local structure studies using X-ray absorption spectroscopy. For probing the local structure, the EXAFS measurements at Th L3 edge have been carried out in transmission mode, while the measurements at Gd L3 edge have been carried out in fluorescence mode. In transmission mode, three ionization chambers (300 mm length each) have been used for data collection, one ionization chamber for measuring incident flux (I0), second one for measuring transmitted flux (It) and the third ionization chamber for measuring EXAFS spectrum of a reference metal foil for energy calibration. Appropriate gas pressure and gas mixture have been chosen to achieve 10–20% absorption in first ionization chamber and 70–90% absorption in second ionization chamber to improve the signal to noise ratio. In case of fluorescence measurements, one ionization chamber is used for measuring the incident flux (I0), while a Si drift detector (Vortex detector) is used in standard 45° geometry to measure the fluorescence signal (If). For transmission measurement, the absorption coefficient μ is obtained using the relation:where, x is the thickness of the absorber.And for fluorescence measurement the absorption coefficient μ is obtained using the relation:
| |
 | (5) |
The normalized EXAFS spectra of Gd doped Th0.7U0.3O2 are shown in Fig. 9 and 10 at Th L3 edge and Gd L3 edge respectively. The insets of the figures are the XANES spectra plotted along with reference sample. The edge position of Th all the samples matches with that of ThO2 standard sample suggesting that Th is in +4 oxidation state in all the samples and there is no change in edge position with variation in Gd concentration. It can also be seen that the edge positions of Gd in all the Gd doped thoria urania samples coincide with that in Gd2O3 suggesting that Gd is present in +3 oxidation state in all the samples. The normalised XANES spectra at U L3 edge for all the samples are shown in Fig. 11 along with UF4 and UO3 (with oxidation state +4 and +6 respectively). It can be seen that uranium edge position for TU, TUG1, TUG2 and TUG3 lies in between UF4 and UO3, which indicates uranium in these samples is present in mixture of +4 and +5/+6 oxidation states. The average uranium valence observed in these samples also showed more than 4 (Table 2) for all the samples corroborates with the XANES results. In order to take care of the oscillations in the absorption spectra μ(E) has been converted to absorption function χ(E) defined as follows51
| |
 | (6) |
where
E0 absorption edge energy,
μ0(
E0) is the bare atom background and Δ
μ0(
E0) is the step in
μ(
E) value at the absorption edge.
 |
| | Fig. 9 Normalised EXAFS spectra of Th0.7U0.3O2 and Gd doped Th0.7U0.3O2 at Th L3 edge. The inset of the figure shows the XANES spectra of the same along with standard ThO2. | |
 |
| | Fig. 10 Normalised EXAFS spectra of Th0.7U0.3O2 and Gd doped Th0.7U0.3O2 at Gd L3 edge. The inset of the figure shows the XANES spectra of the same along with standard Gd2O3. | |
 |
| | Fig. 11 Normalized XANES spectra at U L3 edge for TU, TUG1, TUG2 and TUG3 along with UF4 and UO3. | |
The energy dependent absorption coefficient χ(E) has been converted to the wave number dependent absorption coefficient χ(k) using relation,
| |
 | (7) |
where ‘
m’ is the electron mass.
χ(
k) is weighted by
k2 to amplify the oscillation at high
k and the
χ(
k)
k2 functions are Fourier transformed in
R space to generate the
χ(
R)
versus R spectra in terms of the real distances from the center of the absorbing atom. The set of EXAFS data analysis available in within IFEFFIT software package have been used for EXAFS data analysis.
52 This includes background reduction and Fourier transform to derive the
χ(
R)
versus R spectra from the absorption spectra (using ATHENA software), generation of the theoretical EXAFS spectra starting from an assumed crystallographic structure and finally fitting of experimental data with the theoretical spectra using ARTEMIS software.
The χ(R) versus R spectra have been generated for all the samples from the μ(E) versus E spectra following the methodology described above and are shown in Fig. 12 and 13 at Th L3 edge and Gd L3 edge respectively along with the best fit theoretical simulation. The structural parameters (atomic coordination and lattice parameters) of Th0.7U0.3O2 used for simulation of theoretical EXAFS spectra of the samples have been obtained from XRD results. The bond distances, co-ordination numbers (including scattering amplitudes) and disorder (Debye–Waller) factors (σ2), which give the mean square fluctuations in the distances, have been used as fitting parameters. The best-fitted results are summarized in ESI Tables 2 and 3† at Th L3 edge and Gd L3 edge respectively. In thoria urania solid solution, thorium and uranium is randomly distributed and both thorium and uranium can act as scattering centers. Since the compounds were prepared by taking thorium and uranium as per the compositions reported here and also uranium concentration kept constant in all compositions, three paths, i.e., Th–O, Th–Th from the first coordination sphere and Th–O path from the immediate coordination sphere were considered for fitting the spectra in the case of Th L3 edge and Gd–O, Gd–Gd and Gd–O immediate coordination sphere for fitting the Gd L3 edge. For the present analysis, the Fourier transform range used in k space is 2–11 Å−1 and the fitting range used in case of both Th and Gd edge data is ∼1–4.5 Å.
 |
| | Fig. 12 Fourier transformed EXAFS spectra of (a) Th0.7U0.3O2, (b) Th0.65U0.3Gd0.05O2+y, (c) Th0.60U0.3Gd0.10O2+y and (d) Th0.55U0.3Gd0.15O2+y at Th L3 edge (scatter points) and theoretical fit (solid line). (The spectra shown here is phase uncorrected which shows peak at lower R than actual.) | |
 |
| | Fig. 13 Fourier transformed EXAFS spectra of (a) Th0.65U0.3Gd0.05O2+y, (b) Th0.60U0.3Gd0.10O2+y and (c) Th0.55U0.3Gd0.15O2+y at Gd L3 edge (scatter points) and theoretical fit (solid line). The spectra shown here is phase uncorrected which shows peak at lower R than actual. | |
The Fourier transform EXAFS (FT-EXAFS) spectra of Th L3 edge of Th0.7U0.3O2 shown in Fig. 12 displays two main peaks at 1.9 Å and 3.75 Å along with smaller peaks at 3.1 Å and 4.25 Å. The peaks below 1.5 Å are the background contribution. The first peak at 1.9 Å (Fig. 10) is the contribution of 8 oxygen atoms at 2.38 Å. The second peak at 3.75 Å and small peak at 3.1 Å is contribution of 12 Th/U atoms at distance of 3.91 Å. The peak at 4.25 Å is contribution of 24 oxygen atoms as well as multiple scattering paths. The bond length obtained from EXAFS fitting here is in good agreement with that reported by Hubert et al.53 It can be further observed from ESI Table 2,† that there is no significant change in the Th–O and Th–Th distances upon Gd doping which is as expected since the ionic radii of 8 coordinated Th4+ (1.05 Å) is not so different from ionic radii of 8 coordinated Gd3+ (1.0 Å).
The oxygen co-ordinations in both 1st and 3rd shells in TGU1 and TGU3 samples were decreased significantly compared to Gd free thoria–urania samples which was expected when Th4+ ions were replaced by Gd3+ ions. The maximum increase in the oxygen co-ordination was observed in the case of TUG2 samples. When Gd3+ replaces Th4+ atom in the solid solution, the charge neutralisation may occur via three processes. There may be creation of oxygen vacancies to compensate the charge. Secondly, some of the uranium atoms present in the solid solution may get oxidised to higher oxidation state and compensate the charge valance. Although, these two possibilities alone by themselves seem to be hypothetical, the third possibility of charge compensation by combination of the two possibilities seems more realistic. The O/M results of TGU2 samples showed that the O/M in this sample is slightly enhanced than that of expected value compared to TGU1 and TGU3. This shows that in TGU2 samples the contribution by uranium oxidation is more for charge compensation than by formation of oxygen vacancies. Whereas more oxygen vacancies are prevalent in TGU1 and TGU3 for charge compensation which explains the lesser oxygen coordination in these samples. The heat capacity values of TGU2 samples also could be seen elevated than that of Gd free thoria urania samples throughout the temperature range studied. This higher heat capacity trend in TGU2 sample is attributed to the defects due to the excess oxygen atoms centred around uranium atoms.
The FT-EXAFS spectra of Gd doped Th0.7U0.3O2 samples measured at Gd L3 edge are shown in Fig. 13. It can be seen that the FT-EXAFS spectra are similar for 5% and 15% Gd doped samples but the second and third coordination peaks at 3 Å and 3.6 Å are different for 10% Gd doped sample. The theoretical model for EXAFS fitting at Gd L3 edge is generated by replacing thorium by gadolinium in Th0.7U0.3O2. This theoretical model is able to fit the 5% and 15% Gd doped samples with same number of paths and parameters which are used in EXAFS fitting at Th L3 edge, however the same model could not fit the experimental spectrum of the 10% Gd doped sample satisfactorily. The fitting results are summarised in ESI Table 3† for Gd L3 edge. The first dominant peak of 5% Gd doped Th0.7U0.3O2 at 1.9 Å is a contribution of Gd–O coordination at a distance of 2.38 Å. The second and third peaks at 3 Å and 3.6 Å and small peak at 4.2 Å are contributions of Gd–Th coordination at 3.86 Å, longer Gd–O path at 4.49 Å and multiple scattering paths. However, for the 10% Gd doped sample, the contribution of the longer Gd–O path is found to be insignificant and hence the data have been fitted with a Gd–O and a Gd–Th path only. It can be seen from Table 2 that for the 5% and 15% Gd doped samples, the Gd–O, Gd–Th and longer Gd–O paths are similar to Th–O, Th–Th and longer Th–O path lengths, which suggest that Gd replaces Th in the Th0.7U0.3O2 lattice.
4. Conclusions
(Th0.7−xU0.3Gdx)O2 microspheres where x = 0, 0.05, 0.10 and 0.15 were prepared by internal gelation method. Good quality translucent microspheres were obtained when lower metal concentration and high R was used. The oxides showed positive thermal expansion in the temperature range 298 to 1273 K and the average thermal expansion coefficient decreases with increase in Gd concentration. Specific heat capacities of mixed oxides were measured from 300 to 850 K showed that the temperature dependence of the heat capacities of Gd doped samples is similar to that of un-doped sample. EXAFS measurements at Th L3 edge and Gd L3 edge showed that Gd–O, Gd–Th and longer Gd–O paths are similar to Th–O, Th–Th and longer Th–O path lengths which suggest that Gd replaces Th in the Th0.7U0.3O2 lattice. The higher oxygen coordination in TUG2 samples showed that the charge compensation in these samples were predominantly occurred by oxidation of U4+ to higher oxidation state compared to TUG1 and TUG3 samples.
Acknowledgements
The authors are thankful to Dr S. Kannan, Head, Fuel Chemistry Division for giving valuable guidance and showing keen interest in this work.
References
- R. K. Sinha and A. Kakodkar, Nucl. Eng. Des., 2006, 236, 683–700 CrossRef CAS.
- J. Banerjee, T. R. G. Kutty, A. Kumar, H. S. Kamath and S. Banerjee, J. Nucl. Mater., 2011, 408, 224–230 CrossRef CAS.
- J. S. Herring, P. E. MacDonald, K. D. Weaver and C. Kullberg, Nucl. Eng. Des., 2001, 203, 65–85 CrossRef CAS.
- E. Hellstrand, Trans. Am. Nucl. Soc., 1982, 40, 181–182 Search PubMed.
- F. B. Skogen, L. A. Nielsen and R. G. Grummer, Trans. Am. Nucl. Soc., 1982, 40, 194–198 Search PubMed.
- C. W. Lau, C. Demazière, H. Nylén and U. Sandburg, Prog. Nucl. Energy, 2012, 61, 48–56 CrossRef CAS.
- Advanced Fuel Pellet Materials and Designs for Water Cooled Reactors, IAEA-TECDOC-1416, IAEA, Vienna, 2004 Search PubMed.
- Advanced Fuel Pellet Materials and Fuel Rod Design for Water Cooled Reactors, IAEA-TECDOC-1654, IAEA, Vienna, 2010 Search PubMed.
- K. Hesketh, Burnable poison-doped fuel, in Comprehensive Nuclear Materials, ed. R. J. M. Konings, Elsevier, 2012 Search PubMed.
- K. Une and M. Oguma, J. Nucl. Mater., 1982, 110, 215–222 CrossRef CAS.
- T. B. Lindemer and A. L. Sutton Jr, J. Am. Ceram. Soc., 1988, 71, 553–561 CrossRef CAS.
- M. Peehs, W. Dörr, G. Gradel and G. Maier, J. Nucl. Mater., 1982, 106, 221–230 CrossRef CAS.
- C. Keller, U. Berndt, H. Engerer and L. Leitner, J. Solid State Chem., 1972, 4, 453–456 CrossRef CAS.
- M. Keskar, U. M. Kasar, K. Krishnan, N. D. Dahale, S. K. Sali and S. Kannan, J. Nucl. Mater., 2014, 452, 24–30 CrossRef CAS.
- Characteristics and Use of Urania-Gadolinia Fuels, IAEA-TECDOC-844, IAEA, Vienna, 1995 Search PubMed.
- A. Baena, T. Cardinaels, B. Vos, K. Binnemans and M. Verwerft, J. Nucl. Mater., 2015, 461, 271–281 CrossRef CAS.
- L. N. Grossman, D. R. Packard and H. W. Hill, Colloq. Int. C. N. R. S., 1972, 205, 453–458 Search PubMed.
- R. Yuda and K. Une, J. Nucl. Mater., 1991, 178, 195–203 CrossRef CAS.
- H. G. Riella, M. Durazzo, M. Hirata and R. A. Nogueira, J. Nucl. Mater., 1991, 178, 204–211 CrossRef CAS.
- H. Assmann, M. Peehs and H. Roepenack, J. Nucl. Mater., 1988, 153, 115–126 CrossRef CAS.
- H. Muta, T. Kawano, M. Uno, Y. Ohishi, K. Kurosaki and S. Yamanaka, J. Nucl. Mater., 2013, 434, 124–128 CrossRef CAS.
- M. D. Mathews, B. R. Ambekar and A. K. Tyagi, J. Nucl. Mater., 2005, 341, 19–24 CrossRef CAS.
- H. G. Riella, M. Durazzo, M. Hirata and R. A. Nogueira, J. Nucl. Mater., 1991, 178, 204–211 CrossRef CAS.
- R. Manzel and W. O. Dörr, Am. Ceram. Soc. Bull., 1980, 59, 601–603 CAS.
- S. Fukushima, T. Ohmichi, A. Maeda and H. Watanabe, J. Nucl. Mater., 1982, 105, 201–210 CrossRef CAS.
- C. Miyake, M. Kanamaru and S. Imoto, J. Nucl. Mater., 1986, 138, 142–144 CrossRef CAS.
- A. G. Leyva, D. Vega, V. Trimarco and D. Marchi, J. Nucl. Mater., 2002, 303, 29–33 CrossRef CAS.
- M. Durazzo, F. B. V. Oliveira, E. F. U. de Carvalho and H. G. Riella, J. Nucl. Mater., 2010, 400, 83–188 CrossRef.
- D. Horlait, N. Clavier, N. Dacheux, R. Cavalier and R. Podor, Mater. Res. Bull., 2012, 47, 4017–4025 CrossRef CAS.
- G. Gündüz, I. Uslu, I. Onal, H. H. Durmazucar, T. Ozturk, A. A. Aksit, B. Kopuz, F. Can, S. Can and R. Uzmen, Nucl. Technol., 1995, 111, 63–69 CrossRef.
- G. Gündüz and I. Uslu, J. Nucl. Mater., 1996, 231, 113–120 CrossRef.
- R. V. Pai, S. K. Mukerjee and V. N. Vaidya, J. Nucl. Mater., 2004, 325, 159–168 CrossRef CAS.
- W. Davies and W. Gray, Talanta, 1964, 11, 1203–1211 CrossRef CAS.
- K. D. Singh Mudher, M. Keskar, K. Krishnan and V. Venugopal, J. Alloys Compd., 2005, 396, 275–279 CrossRef.
- V. K. Wadhawan, LATPAR, A Least Squares Program, Neutron Physics Division, Mumbai, India, 1972 Search PubMed.
- K. Hirata, K. Moriya and Y. Waseda, J. Mater. Sci., 1977, 12, 838–839 CrossRef CAS.
- http://www.rrcat.gov.in/technology/accel/srul/beamlines/index.html.
- S. Basu, C. Nayak, A. K. Yadav, A. Agrawal, A. K. Poswal, D. Bhattacharyya, S. N. Jha and N. K. Sahoo, J. Phys.: Conf. Ser., 2014, 493, 12032 CrossRef.
- M. H. Lloyd, K. Bischoff, K. Peng, H. U. Nissen and R. Wessichen, J. Inorg. Chem., 1976, 38, 1141–1147 CAS.
- M. Amaya, K. Une and M. Hirai, J. Nucl. Sci. Technol., 2004, 41, 108–115 CrossRef CAS.
- K. Une, J. Nucl. Sci. Technol., 1986, 23, 1020–1022 CrossRef CAS.
- T. Ohmichi, S. Fukushima, A. Maeda and H. Watanabe, J. Nucl. Mater., 1981, 102, 40–46 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. Venkata Krishnan, G. Panneerselvam, P. Manikandan, M. P. Antony and K. Nagarajan, J. Nucl. Radiochem. Sci., 2009, 10, 19–26 Search PubMed.
- M. D. Mathews, B. R. Ambekar and A. K. Tyagi, J. Nucl. Mater., 2001, 288, 83–85 CrossRef CAS.
- P. T. Dawson and P. C. Walker, Experimental Research in Catalysis Research, ed. R. B. Anderson and T. T. Dawson, Academic press, Newyork, 1976, vol. 3, p. 211 Search PubMed.
- J. J. F. Scholten, A. P. Pijpers and A. M. L. Hustings, Catal. Rev., 1985, 27, 151–206 CrossRef CAS.
- J. W. Jenkins, B. D. McNicol and S. D. Robertson, Chem. Technol. Fuels Oils, 1977, 7, 302–316 Search PubMed.
- N. W. Hurst, S. J. Gentry, A. Jones and B. D. McNicol, Catal. Rev.: Sci. Eng., 1982, 24, 233–309 CrossRef CAS.
- K. Une and M. Oguma, J. Nucl. Mater., 1985, 131, 88–91 CrossRef CAS.
- X-Ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES, ed. D.C. Konigsberger and R. Prince, Wiley, New York, 1988 Search PubMed.
- M. Newville, B. Ravel, D. Haskel, J. J. Rehr, E. A. Stern and Y. Yacoby, Phys. B: Condens. Matter, 1995, 208, 154–156 CrossRef.
- S. Hubert, J. Purans, G. Heisbourg, P. Moisy and N. Dacheux, Inorg. Chem., 2006, 45, 3887–3894 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra07621a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.