
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical synthesis of the hexasaccharide related to the repeating unit of the capsular polysaccharide from carbapenem resistant Klebsiella pneumoniae 2796 and 3264†
Vikramjit
Sarkar
and
Balaram
Mukhopadhyay
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, India. E-mail: sugarnet73@hotmail.com; Tel: +91 9748 261742
First published on 18th April 2016
Abstract
The total synthesis of the hexasaccharide repeating unit of the CPS from carbapenem resistant K. pneumoniae 2796 and 3264 is reported using a sequential glycosylations approach. The total synthesis has been accomplished by glycosylation of rationally protected monosaccharide synthons derived from the commercially available sugars. The required uronic acid on the galactose moiety was successfully installed by a TEMPO-mediated late stage oxidation. The glycosylations were performed by the NIS-mediated activation of thioglycosides using H2SO4-silica as the promoter. Chloroacetate group was extensively used as a temporary protecting group to facilitate stereoselective glycosylations.
Introduction
Bacterial capsular polysaccharides (CPS) and lipopolysaccharides (LPS) are responsible for their virulence factor. CPS and LPS are made up of oligosaccharide repeating units with varied sugar residues. As they remain exposed on the outer surface of the bacterial cell wall, they play pivotal role in infection. Due to the presence of different sugar residues in the oligosaccharide repeats, CPS and LPS demonstrate diverse character and act as the elicitor of innate immune responses. Literature reports suggest that there are potential scope with these bacterial O-antigens as vital candidates for anti-microbial agents and vaccine-targets.1–3 However, it require tedious isolation and purification processes to harness these complex oligosaccharides in adequate quantity. Thus the chemical synthesis of the required oligosaccharides remains the only way to explore their vivid biological roles and potential as vaccine targets.Klebsiella pneumoniae (K. pneumoniae) is an opportunistic pathogen that is responsible for community or hospital acquired infections. It mostly affects the urinary and respiratory tracts. Both CPS and LPS of the K. pneumonia are found to be responsible for the virulence. The CPS antigens are used for K-typing of K. pneumoniae whereas LPS antigens are used for O-typing.4 The capsular antigens are protective against capsular pathogens such as H. influenza type b, meningococci and pneumococci.5 Among large number of K. pneumoniae K-antigens only a few are associated with human disease.6 This is a limiting factor for the development of suitable vaccine against this pathogen. Particularly the carbapenem resistant K-antigens (CRKP) are rarely known in the literature. Only recently, Kubler-Kielb has reported the structures of the CRKP CPS and LPS from clinical isolates collected from the infected patients of a CRKP outbreak in the US.7 Herein, we report the total synthesis of the hexasaccharide repeating unit of the CPS from K. pneumoniae 2796 and 3264 in the form of its p-methoxyphenyl glycoside (Fig. 1). The particular aglycon in the reducing end will enable us to form further glycoconjugates after the selective removal of the same from the per-O-acetylated derivative of the target oligosaccharide.
 | ||
| Fig. 1 Structure of the target hexasaccharide in the form of its p-methoxyphenyl glycoside. | ||
Results and discussion
Judicious retro-synthetic analysis indicated that a sequential glycosylations strategy would be the best fit for the successful synthesis of the target hexasaccharide 1. Thus, three rhamnose moieties were planned to be stitched by using the same synthon 5 with the reducing end galactose moiety 6. The chloroacetate group was thought to be the perfect choice as a temporary protecting group as it can ensure the required 1,2-trans glycosylations as well as can be de-protected selectively to pave the path for introduction of the next sugar unit. Next, a suitably protected galactose synthon 14 having a non-participating group at 2-position thought to be ideal for the required 1,2-cis linkage. A per-O-acylated rhamnose derivative 17 will complete the target hexasaccharide in its protected form. Finally, a TEMPO mediated oxidation of the primary hydroxyl group of the non-reducing end galactose moiety followed by global de-protection would furnish the required molecule (Fig. 2). | ||
| Fig. 2 Retrosynthetic analysis for the total synthesis of the target hexasaccharide 1. | ||
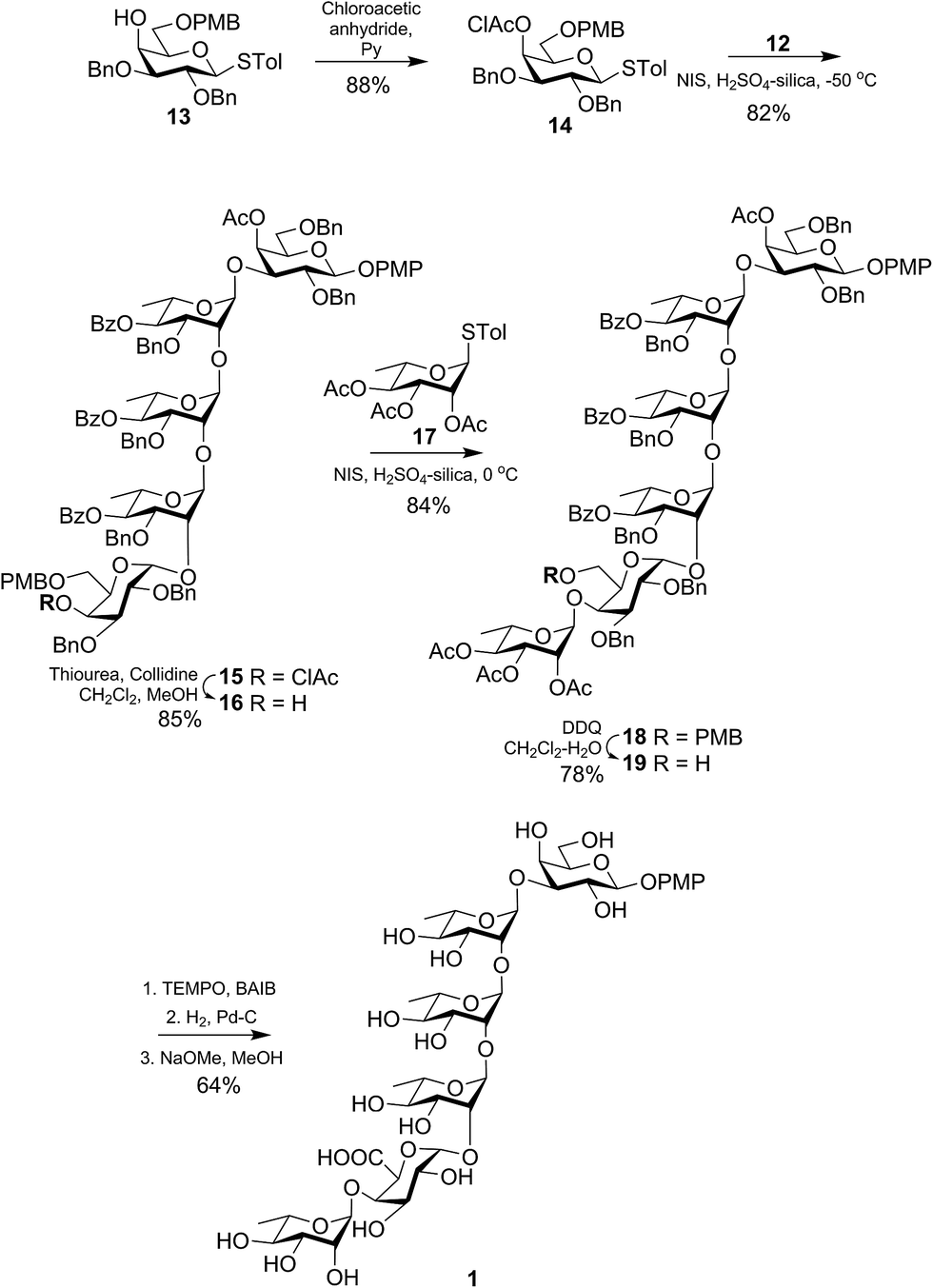
Therefore, the we started our synthesis with known p-tolyl 4-O-benzoyl-2,3-O-isopropylidene-1-thio-α-L-rhamnopyranoside (2).8 Hydrolysis of the isopropylidene group using 80% AcOH at 80 °C (ref. 9) gave the diol 3 in 91% yield. Next, selective benzylation at the equatorial 3-OH was accomplished by following stannylene chemistry10 to give the derivative 4 in 85% yield. Finally, protection of the sole 2-OH with chloroacetate11 group afforded the required donor 5 in 89% yield. Donor 5 was coupled with the known acceptor 6 (ref. 12) by the activation of the thiotolyl using NIS in the presence of H2SO4-silica13 at 0 °C to afford the disaccharide 7 in 91% yield. It is worth noting that the use of H2SO4-silica as the promoter for NIS-mediated activation of the thioglycoside donor found to be beneficial compared to the use of toxic, fuming and hygroscopic TfOH or TMSOTf. Further, selective de-protection of the chloroacetate group using thiourea14 gave the disaccharide acceptor 8 in 87% isolated yield. Subsequently, glycosylations with the same donor 5 followed by de-protection of the chloroacetate group using the same reagent combination and condition was iterated twice to obtain the tetrasaccharide acceptor 12 (Scheme 1). The yields of the individual steps involved are mentioned in the Scheme 1.
 | ||
| Scheme 1 Synthesis of the tetrasaccharide acceptor 12. | ||
In a separate experiment, known p-tolyl 2,3-di-O-benzyl-6-O-(4-methoxybenzyl)-1-thio-β-D-galactopyranoside (13)15 was treated with chloroacetic anhydride in the presence of dry pyridine to give the completely protected donor (14) in 88% yield. Next, glycosylations of the donor 14 with the tetrasaccharide acceptor 12 using NIS in the presence of H2SO4-silica at −50 °C gave the protected pentasaccharide 15 in 82% yield. Presence of the non-participating benzyl group at the 2-position of the galactosyl donor 14 and the reaction at very low temperature assured the formation of the desired 1,2-glycoside as the sole isolated product. Further, selective de-protection of the chloroacetate group using thiourea afforded the pentasaccharide acceptor 16 in 85% yield. Finally, glycosylations of 16 with the known donor 17 (ref. 16) using the same NIS/H2SO4-silica at 0 °C furnished the protected hexasaccharide 18 in 84% yield. At this stage, the strategically placed 4-methoxybenzyl group was selectively de-protected by oxidative cleavage using DDQ17 to afford the hexasaccharide derivative 19 in 78% isolated yield. Oxidation of the primary hydroxyl group using TEMPO in the presence of bis-acetoxy iodobenzene (BAIB)18 followed by catalytic hydrogenolysis and Zemplen de-O-acetylation19 gave the target hexasaccharide 1 in 64% yield over three steps (Scheme 2). The amorphous white powder of compound 1 was triturated with CH2Cl2 and filtered to remove aromatic impurities.
 | ||
| Scheme 2 Synthesis of the target hexasaccharide 1. | ||
Conclusions
In conclusion, we have successfully accomplished the total synthesis of the hexasaccharide repeating unit of the CPS from K. pneumoniae 2796 and 3264 in the form of its p-methoxyphenyl glycoside. The practical synthetic strategy used the minimum protecting group manipulations and the chloroacetate group was used extensively as the temporary protecting group to ensure stereoselective glycosylations using rhamnose synthons. A TEMPO-mediated late stage oxidation was used successfully to generate the desired uronic acid moiety. The synthetic will definitely enhance the scope for further biological evaluation of the target oligosaccharide related to the carbopenem resistant K. pneumoniae strains and pave the path for a potential vaccine target.Experimental section
General methods
All solvents and reagents were dried prior to use according to standardized methods.20 The commercially purchased reagents were used without any further purification unless mentioned otherwise. All reactions were monitored by Thin Layer Chromatography (TLC) on Silica-Gel 60-F254 with detection by fluorescence followed by charring after immersion in 10% ethanolic solution of H2SO4. Flash chromatography was performed with Silica Gel 230–400 mesh. 1H and 13C NMR spectra were recorded on Bruker Avance 500 MHz spectrometer (1H NMR at 500 MHz and 13C NMR at 125 MHz). HRMS analysis was performed with Micromass Q-TOF micro (Waters Corporation) instrument by +ve mode electro-spray ionization.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 36 mL) and the solution was stirred at 80 °C for 2 h until the starting material was completely converted to a slower moving spot as suggested by TLC (n-hexane–EtOAc; 3:1). The solvents were evaporated and co-evaporated twice with toluene followed by purification of the crude product by flash chromatography using (n-hexane–EtOAc; 3.5:1) to afford pure compound 3 (4.9 g, 91%) as colourless syrup. [α]25D +103° (c 1.0, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.09–7.15 (m, 9H, ArH), 5.51 (d, 1H, J1,2 1.5 Hz, H-1), 5.09 (t, 1H, J3,4, J4,5 9.5 Hz, H-4), 4.50 (m, 1H, H-5), 4.26 (bd, 1H, J1,2 1.5 Hz, H-2), 4.07 (dd, 1H, J2,3 3.0 Hz, J3,4 9.5 Hz, H-3), 3.31 (bs, 1H, OH), 2.79 (bs, 1H, OH), 2.34 (s, 3H, SC6H4CH3), 1.31 (d, 3H, J5,6 6.0 Hz). 13C NMR (CDCl3, 125 MHz) δ: 167.7 (COC6H5), 137.9, 133.6 (2), 132.1 (2), 129.9 (4), 129.3, 128.5 (2) (ArC), 87.6 (C-1), 76.6, 72.3, 70.9, 67.0, 21.1 (SC6H4CH3), 17.5 (C–CH3). HRMS calcd for C20H22O5SNa (M + Na)+: 397.1086, found: 397.1084.
:1) as eluent to give the pure compound 4 (5.0 g, 85%). [α]25D +96° (c 1.0, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.03–7.10 (m, 14H, ArH), 5.45 (d, 1H, J1,2 < 1.0 Hz, H-1), 5.42 (t, 1H, J3,4, J4,5 9.5 Hz, H-4), 4.65, 4.51 (ABq, 2H, JA–B 12.0 Hz, CH2Ph), 4.37 (m, 1H, H-5), 4.32 (dd, 1H, J1,2 < 1.0 Hz, J2,3 3.0 Hz, H-2), 3.90 (dd, 1H, J2,3 3.0 Hz, J3,4 9.5 Hz, H-3), 3.08 (bs, 1H, OH), 2.31 (s, 3H, S–C6H4–CH3), 1.24 (d, 3H, J5,6 6.5 Hz). 13C NMR (CDCl3, 125 MHz) δ: 165.9 (COC6H5), 137.9, 137.3, 133.3, 132.1 (2), 130.0 (3), 129.9 (2), 128.6 (2), 128.5 (2), 128.1 (4) (ArC), 87.4 (C-1), 77.5, 73.2, 71.7, 69.8, 67.7, 21.2 (SC6H4CH3), 17.5 (C–CH3). HRMS calcd for C27H22O5SNa (M + Na)+: 487.1555, found: 487.1553.
:1) showed complete conversion of the reactant to a faster moving spot, the mixture was evaporated in vacuo and co-evaporated twice with toluene to obtain a syrupy residue, which was then purified by flash chromatography using n-hexane–EtOAc (8:1) as eluent to give the pure compound 5 (5.2 g, 89%) as light yellow syrup. [α]25D +113° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.04–7.15 (m, 14H, ArH), 5.73 (dd, 1H, J1,2, 1.5 Hz, J2,3 3.0 Hz, H-2), 5.45 (d, 1H, J1,2 1.5, H-1), 5.36 (t, 1H, J3,4, J4,5 10.0 Hz, H-4), 4.66, 4.45 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.36 (m, 1H, H-5), 4.25, 4.17 (ABq, 2H, JA–B 15.5 Hz, COCH2Cl), 3.98 (dd, 1H, J2,3 3.0 Hz, J3,4 10.0 Hz, H-3), 2.35 (s, 3H, S–C6H4–CH3), 1.29 (d, 3H, J5,6 6.5 Hz). 13C NMR (CDCl3, 125 MHz) δ: 166.7 (COCH2Cl), 165.5 (COC6H5), 138.3, 136.8, 133.2, 132.4 (2), 129.9 (2), 129.8 (2), 129.5, 129.1, 128.3 (2), 128.2 (2), 128.0 (2), 127.8 (ArC), 86.1 (C-1), 74.2, 72.6, 71.8, 71.3, 67.9, 74.2, 72.6, 71.8, 71.3, 67.9, 40.8 (COCH2Cl), 21.0 (SC6H4CH3), 17.2 (C–CH3). HRMS calcd for C29H29ClO6SNa (M + Na)+: 563.1271, found: 563.1269.
:1) indicated complete consumption of the donor. The reaction mixture was neutralized with Et3N and the mixture was filtered through a pad of Celite. The filtrate was washed successively with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). Organic layer was separated, dried over Na2SO4 and evaporated in vacuo. The syrupy crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (4:1) as eluent to afford pure disaccharide 7 (2.3 g, 91%) as white foam. [α]25D +86° (c 0.9, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.07–7.12 (m, 20H, ArH), 7.05, 6.82 (2d, 4H, C6H4OCH3), 5.52 (dd, 1H, J2′,3′ 1.5 Hz, J1′,2′ < 1.0 Hz, H-2′), 5.39 (bd, 1H, J3,4 1.5 Hz, H-4), 5.23 (d, 1H, J1′,2′ < 1.0 Hz, H-1′), 5.22 (t, 1H, J3′,4′, J4′,5′ 10.0 Hz, H-4′), 5.06, 4.75 (ABq, 2H, JA–B 11.0 Hz, CH2Ph), 4.92 (d, 1H, J1,2 7.0 Hz, H-1), 4.61, 4.42 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.55, 4.48 (ABq, 2H, JA–B 12.0 Hz, CH2Ph), 4.21, 4.12 (ABq, 2H, JA–B 15.5 Hz, CH2Cl), 4.13 (m, 1H, H-5′), 3.93 (m, 2H, H-2, H-3), 3.88 (t, 1H, J4,5, J5,6a, J5,6b 6.5 Hz, H-5), 3.83 (dd, 1H, J2′,3′ 1.5 Hz, J3′,4′ 10.0 Hz, H-3′), 3.78 (s, 3H, OC6H4OCH3), 3.58 (m, 2H, H-6a, H-6b), 1.97 (s, 3H, COCH3), 1.25 (d, 3H, J5′,6′ 6.5 Hz). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 166.6 (COCH2Cl), 165.6 (COC6H5), 155.4, 151.1, 137.6, 137.5, 137.3, 133.1, 129.9 (2), 129.7, 128.5 (4), 128.3 (4), 128.2 (2), 127.8 (4), 127.7 (2), 127.6, 118.2 (2), 114.6 (2) (ArC), 102.9 (C-1), 98.5 (C-1′), 79.0, 75.1, 74.7, 73.6, 73.5, 72.7, 72.5, 71.1, 69.9, 69.1, 68.2, 67.3, 55.6 (OC6H4OCH3), 40.8 (COCH2Cl), 20.6 (COCH3), 17.6 (C–CH3). HRMS calcd for C51H53ClO14Na (M + Na)+: 947.3022, found: 947.3019.
:3) for 10 hours when TLC (n-hexane–EtOAc; 2.5:1) confirmed the complete conversion of the starting material to a slower moving spot. The solvents were evaporated in vacuo, the solid residue was dissolved in CH2Cl2 and washed with 1 (N) HCl (2 × 30 mL). The organic layer was collected, filtered, dried (Na2SO4) and evaporated in vacuo. The crude residue thus obtained was further subjected to flash chromatography using n-hexane–EtOAc (3:1) as eluent to give the pure disaccharide acceptor 8 (1.8 g, 87%). [α]25D +121° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.09–7.17 (m, 20H, ArH), 7.07, 6.83 (2d, 4H, C6H4OCH3), 5.42 (m, 1H, H-4), 5.34 (t, 1H, J3′,4′, J4′,5′ 9.5 Hz, H-4′), 5.30 (d, 1H, J1′,2′ < 1 Hz, H-1′), 5.04, 4.78 (ABq, 2H, JA–B 11.0 Hz, CH2Ph), 4.93 (d, 1H, J1,2 7.5 Hz, H-1), 4.61, 4.50 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.56, 4.49 (ABq, 2H, JA–B 11.5 Hz, CH2Ph), 4.10 (m, 1H, H-5′), 3.97–3.89 (m, 4H, H-2, H-3, H-5, H-2′), 3.78 (s, 3H, OC6H4OCH3), 3.73 (dd, 1H, J2′,3′ 2.0 Hz, J3′,4′ 9.5 Hz, H-3′), 3.60 (m, 2H, H-6a, H-6b), 2.60 (bs, 1H, OH), 2.03 (s, 3H, COCH3), 1.26 (d, 3H, J5′,6′ 6.5 Hz). 13C NMR (CDCl3, 125 MHz) δ: 169.8 (COCH3), 165.7 (COC6H5), 155.4, 151.3, 137.8, 137.7, 137.4, 133.0, 129.9, 129.8, 128.4 (2), 128.3 (4), 128.3 (3), 128.2 (2), 127.8, 127.7 (2), 127.7 (2), 127.6 (2), 118.3 (2), 114.5 (2) (ArC), 102.9 (C-1), 100.5 (C-1′), 79.0, 75.8, 75.4, 75.0, 73.6, 72.9, 72.8, 71.3, 69.9, 68.4, 68.2, 66.9, 55.5 (OC6H4OCH3), 20.6 (COCH3), 17.5 (C–CH3). HRMS calcd for C49H52O13Na (M + Na)+: 871.3306, found: 871.3304.
:1) confirmed the complete consumption of the donor. Then the reaction mixture was filtered through a pad of Celite. The filtrate was washed successively with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). Organic layer was separated, dried (Na2SO4) and evaporated in vacuo. The crude product thus obtained was subjected to flash chromatography using n-hexane–EtOAc (3:1) as the eluent to obtain the pure trisaccharide 9 (2.1 g, 90%) as white foam. [α]25D +87° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.14–7.12 (m, 30H, ArH), 7.01, 6.79 (2d, 4H, C6H4OCH3), 5.64 (dd, 1H, J2′′,3′′ 2.5 Hz, J1′′,2′′ < 1.0 Hz, H-2′′), 5.41 (m, 1H, H-4), 5.36 (t, 1H, J3′,4′, J4′,5′ 9.5 Hz, H-4′), 5.22 (d, 1H, J1′,2′ < 1.0 Hz, H-1′), 5.21 (t, 1H, J3′′,4′′, J4′′,5′′ 10.0 Hz, H-4′′), 4.98, 4.82 (ABq, 2H, JA–B 11.5 Hz, CH2Ph), 4.89 (m, 2H, H-1, H-1′′), 4.65–4.47 (m, 6H, 3 × CH2Ph), 4.20, 4.12 (ABq, 2H, JA–B 15.0 Hz, COCH2Cl), 4.08 (m, 1H, H-5′), 4.02 (dd, 1H, J2′′,3′′ 2.5 Hz, J3′′,4′′ 10.0 Hz, H-3′′), 3.97 (m, 1H, H-5′′), 3.89–3.86 (m, 4H, H-2, H-2′, H-3, H-5), 3.81 (dd, 1H, J2′,3′ 2.5 Hz, J3′,4′ 9.5 Hz, H-3′), 3.77 (s, 3H, OC6H4OCH3), 3.58 (m, 2H, H-6a, H-6b), 2.03 (s, 3H, COCH3), 1.30 (d, 3H, J5′,6′ 7.0 Hz, C–CH3), 1.07 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.0 (COCH3), 166.4 (COCH2Cl), 165.7 (COC6H5), 165.6 (COC6H5), 155.4, 151.2, 137.8, 137.7, 137.3, 133.1, 130.1, 129.9 (2), 129.8, 128.5 (4), 128.4 (5), 128.3 (5), 128.2 (2), 128.1 (2), 127.8 (4), 127.7, 127.6 (3), 118.3 (3), 114.6 (3) (ArC), 103.0 (C-1), 100.4 (C-1′), 99.4 (C-1′′), 78.3, 76.5, 76.0, 75.5, 74.6, 74.7, 73.5, 73.3, 72.9, 72.5, 71.0, 71.2, 70.3, 69.6, 68.4, 67.6, 67.4, 55.6 (OC6H4OCH3), 40.9 (COCH2Cl), 20.7 (COCH3), 17.8, 17.4 (2 × C–CH3). HRMS calcd for C71H73ClO19Na (M + Na)+: 1287.4332, found: 1287.4330.
:3). The mixture was refluxed for 10 h when the complete conversion of the starting material to a slower moving spot was judged by the TLC (n-hexane–EtOAc; 2:1). The mixture was evaporated in vacuo. The solid residue thus obtained was dissolved in CH2Cl2 and washed with 1 (N) HCl (2 × 30 mL) and brine (2 × 30 mL). The organic layer was separated, dried (Na2SO4) and evaporated in vacuo to get the crude product. It was purified by flash chromatography using n-hexane–EtOAc (5:2) as the eluent to get the pure compound 10 (1.7 g, 88%) as white powder. [α]25D +78° (c 0.7, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.08–7.10 (m, 30H, ArH), 6.96, 6.74 (2d, 4H, C6H4OCH3), 5.35 (m, 1H, H-4), 5.33 (t, 1H, J3′,4′, J4′,5′ 10.0 Hz, H-4′), 5.30 (t, 1H, J3′′,4′′, J4′′,5′′ 10.0 Hz, H-4′′), 5.19 (d, 1H, J1′,2′ 2.0 Hz, H-1′), 5.09 (d, 1H, J1′′,2′′ 2.0 Hz, H-1′′), 4.91, 4.82 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.86 (d, 1H, J1,2 7.5 Hz, H-1), 4.64, 4.52 (ABq, 2H, JA–B 12.0 Hz, CH2Ph), 4.56–4.42 (m, 4H, 2 × CH2Ph), 4.31 (dd, 1H, J1′′,2′′ 2.0 Hz, J2′′,3′′ 3.0 Hz, H-2′′), 4.03 (m, 1H, H-5′), 3.97 (dd, 1H, J1′,2′ 2.0 Hz, J2′,3′ 3.0 Hz, H-2′), 3.93 (m, 1H, H-5′′), 3.91 (dd, 1H, J2′′,3′′ 3.0 Hz, J3′′,4′′ 10.0 Hz, H-3′′), 3.85–3.82 (m, 3H, H-2, H-3, H-5), 3.75 (dd, 1H, J2′,3′ 3.0 Hz, J3′,4′ 10.0 Hz, H-3′), 3.69 (s, 3H, OC6H4OCH3), 3.53 (m, 2H, H-6a, H-6b), 2.62 (bs, 1H, OH), 1.93 (s, 3H, COCH3), 1.23 (d, 3H, J5′,6′ 6.5 Hz, C–CH3), 1.03 (d, 3H, J5′′,6′′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 165.7, 165.6 (2 × COC6H5), 155.3, 151.1, 137.7 (3), 137.3, 133.0, 132.9, 130.0, 129.9, 129.8 (2), 129.7 (2), 128.4 (4), 128.3 (6), 128.2 (4), 128.1 (2), 127.8, 127.7 (4), 127.6, 127.5 (2), 118.2 (2), 114.5 (2) (ArC), 103.0 (C-1), 101.0 (C-1′′), 100.6 (C-1′), 77.8, 75.9 (2), 75.6, 75.2, 74.4, 73.6, 73.2, 72.8 (2), 71.6, 71.3, 69.6, 68.4, 68.0, 67.5, 66.9, 55.5, 20.6 (COCH3), 17.7, 17.3 (2 × C–CH3). HRMS calcd for C69H72O18Na (M + Na)+: 1211.4616, found: 1211.4613.
:1) showed complete consumption of the donor. The mixture was filtered through a Celite pad and the filtrate was successively washed with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). The organic layer was collected, dried (Na2SO4) and evaporated in vacuo. Crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (2:1) as the eluent. The pure tetrasaccharide 11 (1.8 g, 89%) was obtained as white foam. [α]25D +142° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.10–7.10 (m, 40H, ArH), 6.99, 6.78 (2d, 4H, C6H4OCH3), 5.65 (dd, 1H, J1′′′,2′′′ < 1.0 Hz, J2′′′,3′′′ 3.0 Hz, H-2′′′), 5.39–5.34 (m, 3H, H-4, H-4′, H-4′′), 5.22 (d, 1H, J1′,2′ 1.5 Hz, H-1′), 5.21 (t, 1H, J3′′′,4′′′, J4′′′,5′′′ 10.0 Hz, H-4′′′), 5.14 (d, 1H, J1′′,2′′ 1.5 Hz, H-1′′), 4.96, 4.82 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.94 (d, 1H, J1′′′,2′′′ < 1.0 Hz, H-1′′′), 4.88 (d, 1H, J1,2 7.0 Hz, H-1), 4.70–4.42 (m, 8H, 4 × CH2Ph), 4.29 (dd, 1H, J1′′,2′′ 1.5 Hz, J2′′,3′′ < 1.0 Hz, H-2′′), 4.16, 4.09 (ABq, 2H, JA–B 15.5 Hz, COCH2Cl), 4.07–4.01 (m, 4H, H-3′′, H-3′′′, H-5′, H-5′′), 3.96 (m, 1H, H-5′′′), 3.95 (dd, 1H, J1′,2′ 1.5 Hz, J2′,3′ 2.5 Hz, H-2′), 3.90–3.84 (m, 3H, H-2, H-3, H-5), 3.78 (dd, 1H, J2′,3′ 2.5 Hz, J3′,4′ 9.5 Hz, H-3′), 3.76 (s, 3H, OC6H4OCH3), 3.57 (m, 2H, H-6a, H-6b), 1.97 (s, 3H, COCH3), 1.29 (d, 3H, J5′,6′ 6.5 Hz, C–CH3), 1.14 (d, 3H, J5′′,6′′ 6.5 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 166.4 (COCH2Cl), 165.8, 165.7, 165.6 (3 × COC6H5), 155.5, 151.3, 137.8, 137.7, 137.4, 133.2, 133.1 (2), 130.0 (2), 129.9 (2), 129.8 (2), 128.4 (14), 128.3 (8), 128.2 (2), 128.1 (2), 127.8 (6), 127.7 (2), 127.5 (2), 118.3 (2), 114.6 (2) (ArC), 103.1 (C-1), 101.1 (C-1′′), 100.7 (C-1′), 99.2 (C-1′′′), 78.2, 76.1, 75.7, 75.4, 74.6, 73.9, 73.7 (2), 73.3, 73.1, 72.9, 72.7, 71.8 (2), 71.3, 70.2, 69.6, 68.4, 67.7, 67.6, 67.3, 55.6, 40.9 (COCH2Cl), 20.6 (COCH3), 17.8, 17.7, 17.5 (3 × C–CH3). HRMS calcd for C91H93ClO24Na (M + Na)+: 1627.5643, found: 1627.5641.
:3), collidine (0.7 mL, 5.3 mmol) was added and the mixture was refluxed for 12 h till TLC (n-hexane–EtOAc; 2:1) indicated the complete conversion of the starting material to a slower moving spot. The reaction mixture was evaporated in vacuo and the residue was dissolved in CH2Cl2. It was further washed with 1 (N) HCl (2 × 30 mL) and brine (2 × 30 mL). Resulting organic layer was collected, dried (Na2SO4) and evaporated in vacuo to obtain the crude product. It was further purified by flash chromatography using n-hexane–EtOAc (2:1) as the eluent to obtain the pure tetrasaccharide acceptor 12 (1.5 g, 90%) as white foam. [α]25D +102° (c 0.9, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.11–7.12 (m, 40H, ArH), 6.99, 6.79 (2d, 4H, C6H4OCH3), 5.41–5.33 (m, 4H, H-4, H-4′, H-4′′, H-4′′′), 5.23 (d, 1H, J1′,2′ 1.5 Hz, H-1′), 5.19 (d, 1H, J1′′′,2′′′′ 1.5 Hz, H-1′′′), 5.12 (d, 1H, J1′′,2′′ 1.5 Hz, H-1′′), 4.95, 4.82 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.88 (d, 1H, J1,2 7.0 Hz, H-1), 4.72–4.44 (m, 8H, 4 × CH2Ph), 4.37 (dd, 1H, J1′′′,2′′′ 1.5 Hz, J2′′′,3′′′ < 1.0 Hz, H-2′′′), 4.30 (dd, 1H, J1′′,2′′ 1.5 Hz, J2′′,3′′ < 1.0 Hz, H-2′′), 4.08–4.02 (m, 4H, H-3′′, H-3′′′, H-5′, H-5′′), 4.01–3.96 (m, 2H, H-2′, H-5′′′), 3.88–3.84 (m, 3H, H-2, H-3, H-5), 3.79 (dd, 1H, J2′,3′ 3.5 Hz, J3′,4′ 6.5 Hz, H-3′), 3.77 (s, 3H, OC6H4OCH3), 3.58 (m, 2H, H-6a, H-6b), 2.57 (bs, 1H, OH), 1.96 (s, 3H, COCH3), 1.28 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.16 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.11 (d, 3H, J5′′′,6′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.4 (COCH3), 165.9, 165.8, 165.6 (3 × COC6H5), 155.4, 155.2, 127.8, 137.7 (4), 137.5, 133.1 (2), 133.0 (2), 130.6 (6), 129.8 (2), 128.4 (6), 128.3 (4), 128.2 (2), 128.0 (4), 127.8 (6), 127.7 (4), 127.6 (4), 118.3 (2), 114.5 (2) (ArC), 103.0 (C-1), 101.2 (C-1′′′), 100.9 (C-1′′), 100.6 (C-1′), 78.1, 76.4, 76.2, 76.0, 75.4 (2), 74.8, 74.6, 73.7, 73.2, 73.1, 73.0, 72.9, 71.7, 71.6, 71.5, 69.6, 68.5, 68.2, 67.6 (2), 66.9, 55.6, 20.6 (COCH3), 17.8, 17.7, 17.4 (3 × CH3). HRMS calcd for C89H92O23Na (M + Na)+: 1551.5927, found: 1551.5925.
:1) showed complete conversion of the starting material to a faster moving spot. The reaction mixture was evaporated in vacuo and co-evaporated with toluene. The crude product thus obtained was further subjected to purification by flash chromatography using n-hexane–EtOAc (6:1) as eluent to get the pure compound 14 (1.3 g, 88%). [α]25D +139° (c 1.0, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 7.54–6.96 (m, 18H, ArH), 5.74 (dd, 1H, J3,4 3.0 Hz, J4,5 < 1.0 Hz, H-4), 4.83–4.53 (m, 4H, 2 × CH2Ph), 4.67 (d, 1H, J1,2 9.0 Hz, H-1), 4.55, 4.42 (ABq, 2H, JA–B 12.5 Hz, CH2PhOMe), 4.10, 4.03 (ABq, 2H, JA–B 15.5 Hz, CH2Cl), 3.85 (s, 3H, OCH2C6H4OCH3), 3.79 (dd, 1H, J5,6a 6.5 Hz, J6a,6b 9.5 Hz, H-6a), 3.71 (dd, 1H, J2,3 9 Hz, J3,4 3 Hz, H-3), 3.65 (t, 1H, J1,2, J2,3 9.0 Hz, H-2), 3.64 (m, 1H, H-5), 3.55 (dd, 1H, J5,6b 7.5 Hz, J6a,6b 9.5 Hz, H-6b), 2.37 (s, 3H, S–C6H4–CH3). 13C NMR (CDCl3, 125 MHz) δ: 166.6 (COCH2Cl), 159.3, 138.0, 137.6, 137.3, 132.6 (2), 129.7 (2), 129.5 (2), 129.4, 129.3, 128.3 (2), 128.2 (4), 128.0 (2), 127.8, 127.7, 113.7 (2) (ArC), 87.8 (C-1), 80.9, 76.5, 75.6, 75.2, 73.1, 72.0, 68.7, 67.0, 55.1, 40.7 (COCH2Cl), 21.0 (SC6H4CH3). HRMS calcd for C37H39ClO7Na (M + Na)+: 685.2001, found: 685.1998.
:2) suggested the full consumption of the donor, the reaction was quenched by Et3N. It was then followed by filtration of the mixture through a Celite pad. Resulting solution was then successively washed with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). It was then dried (Na2SO4) and evaporated in vacuo. The crude product was then purified by flash chromatography using n-hexane–EtOAc (3:1) as the eluent. Thus the pure pentasaccharide 15 (1.4 g, 82%) was furnished as white foam. [α]25D +68° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.10–6.99 (m, 55H, ArH), 6.81, 6.79 (2d, 4H, C6H4OCH3), 5.69 (bd, 1H, J3,4 1.5 Hz, H-4), 5.42–5.37 (m, 4H, H-4, H-4′, H-4′′, H-4′′′), 5.21 (d, 1H, J1′,2′ 1.0 Hz, H-1′), 5.15 (d, 1H, J1′′′,2′′′′ < 1.0 Hz, H-1′′′), 5.11 (d, 1H, J1′′,2′′ 1.0 Hz, H-1′′), 4.96, 4.83 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.88 (d, 1H, J1,2 7.0 Hz, H-1), 4.83, 4.76 (ABq, 2H, JA–B 10.5 Hz, CH2Ph), 4.79 (d, 1H, J1′′′′,2′′′′ 3.5 Hz, H-1′′′′), 4.70–4.15 (m, 12H, 6 × CH2Ph), 4.53 (m, 1H, H-5′′′′), 4.35 (m, 2H, H-2′′, H-2′′′), 4.12 (m, 1H, H-3′′′′), 4.08–4.01 (m, 3H, H-5′, H-3′′, H-3′′′), 4.01, 4.90 (ABq, 2H, JA–B 15.0 Hz, COCH2Cl), 3.99–3.94 (m, 3H, H-2′, H-5′′, H-5′′′), 3.87–3.86 (m, 2H, H-3, H-5), 3.81–3.78 (m, 2H, H-2, H-3′), 3.77 (s, 3H, OC6H4OCH3), 3.73 (s, 3H, OCH2C6H4OCH3), 3.64 (dd, 1H, J1′′′′,2′′′′ 3.5 Hz, J2′′′′,3′′′′ 6.5 Hz, H-2′′′′), 3.58 (m, 2H, H-6a, H-6b), 3.27 (m, 2H, H-6a′′′′, H-6b′′′′), 1.95 (s, 3H, COCH3), 1.28 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.16 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 166.7 (COCH2Cl), 165.8, 165.6 (2) (3 × COC6H5), 159.1, 155.4, 151.2, 138.8, 138.0, 137.9, 137.7 (2), 137.5, 133.1, 132.9, 130.1, 130.0, 129.9 (2), 129.8, 129.7 (2), 128.4 (10), 128.2 (10), 128.2 (10), 128.1 (4), 127.8 (4), 127.7 (2), 127.6, 127.5 (2), 127.4 (4), 127.3, 118.3 (2), 114.5 (2), 113.6 (2) (ArC), 103.0 (C-1), 101.3 (C-1′′′), 100.6 (C-1′), 99.1 (C-1′′), 97.2 (C-1′′′′), 78.1, 76.0, 75.9, 75.7, 75.5, 75.2, 75.0, 74.7, 74.6, 74.3, 73.7 (2), 73.2, 73.1, 73.0, 72.9, 72.1 (2), 71.6, 71.0, 70.3, 70.0, 68.5, 67.6 (2), 67.4, 67.2, 55.6, 55.1, 41.0 (COCH2Cl), 20.6 (COCH3), 17.8, 17.6 (2) (3 × CH3). HRMS calcd for C119H123ClO30Na (M + Na)+: 2089.7685, found: 2089.7683.
:3) was refluxed for 10 h. When the TLC (n-hexane–EtOAc; 2:1) suggested the complete conversion of the starting material to a slower moving spot, the reaction mixture was evaporated in vacuo. It was then dissolved in CH2Cl2 and washed with brine (2 × 30 mL). The organic layer was collected, dried (Na2SO4) and evaporated. The crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (2:1) to give the pure pentasaccharide acceptor 16 (1.1 g, 85%) as white foam. [α]25D +108° (c 0.7, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.07–6.98 (m, 54H, ArH), 6.78, 6.76 (2d, 4H, C6H4OCH3), 5.44–5.34 (m, 4H, H-4, H-4′, H-4′′, H-4′′′), 5.21 (d, 1H, J1′,2′ 1.0 Hz, H-1′), 5.15 (d, 1H, J1′′′,2′′′ < 1.0 Hz, H-1′′′), 5.11 (d, 1H, J1′′,2′′ 1.0 Hz, H-1′′), 4.95, 4.81 (ABq, 2H, JA–B 11.5 Hz, CH2Ph), 4.87 (d, 1H, J1,2 7.0 Hz, H-1), 4.82 (d, 1H, J1′′′′,2′′′′ 3.0 Hz, H-1′′′′), 4.70–4.40 (m, 14H, 7 × CH2Ph), 4.37–4.36 (m, 3H, H-2′′, H-2′′′, H-4′′′′), 4.13 (m, 1H, H-5′′′′), 4.06–3.90 (m, 8H, H-2′, H-3′′, H-3′′′, H-5′, H-5′′, H-5′′′, H-6a′′′′, H-6b′′′′), 3.80–3.79 (m, 4H, H-2, H-3, H-3′, H-5), 3.77 (s, 3H, OC6H4OCH3), 3.70 (s, 3H, OCH2C6H4OCH3), 3.58 (m, 3H, H-6a, H-6b, H-2′′′′), 3.64 (m, 1H, H-3′′′′), 2.82 (br s, 1H, OH), 1.94 (s, 3H, COCH3), 1.27 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.16 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 165.7 (2), 165.6 (3 × COC6H5), 159.1, 155.5, 151.2, 139.0, 138.0, 137.8 (2), 137.7, 137.6, 133.1, 132.8, 130.2, 130.1 (2), 129.9 (2), 129.8 (4), 129.4 (2), 128.4 (10), 128.3 (4), 128.2 (8), 128.1 (6), 127.9 (4), 127.8 (4), 127.7 (2), 127.6 (2), 127.5 (2), 127.3 (2), 118.3 (2), 114.6 (2), 113.7 (2) (ArC), 103.0 (C-1), 101.3 (C-1′′′), 100.7 (C-1′), 99.2 (C-1′′), 96.8 (C-1′′′′), 78.1, 76.0, 75.5, 74.6, 73.7 (2), 73.3 (4), 73.2 (2), 73.0, 72.7, 72.4, 72.2, 71.6, 70.8, 69.6, 69.4, 68.5, 68.3, 68.2 (2), 67.7, 67.6 (2), 55.6, 55.1, 20.6 (COCH3), 17.8, 17.7, 17.6 (3 × CH3). HRMS calcd for C117H122O29Na (M + Na)+: 2013.7969, found: 2013.7967.
:1) suggested that the whole of the acceptor was consumed, the mixture was neutralised with Et3N and filtered through a Celite pad. The filtrate was successively washed with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). The organic layer was separated, dried (Na2SO4) and evaporated in vacuo to get the crude product. It was purified by flash chromatography using n-hexane–EtOAc (3:2) as eluent to obtain the pure hexasaccharide 18 (960 mg, 84%) as the white foam. [α]25D +132° (c 0.7, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.11–7.00 (m, 54H, ArH), 6.99, 6.79 (2d, 4H, C6H4OCH3), 5.50 (s, 1H, H-2′′′′′), 5.44–5.39 (m, 4H, H-4, H-4′, H-4′′, H-4′′′), 5.37–5.34 (m, 2H, H-3′′′′′, H-5′′′′′), 5.21 (m, 2H, H-1′, H-1′′′′′), 5.09 (m, 2H, H-1′′, H-1′′′), 5.03 (t, 1H, J3′′′′′,4′′′′′, J4′′′′′,5′′′′′ 10.0 Hz, H-4′′′′′), 4.96, 4.83 (ABq, 2H, JA–B 11.5 Hz, CH2Ph), 4.89 (d, 1H, J1,2 6.5 Hz, H-1), 4.78–4.28 (m, 14H, 7 × CH2Ph), 4.70 (d, 1H, J1′′′′,2′′′′ 3.0 Hz, H-1′′′′), 4.36–4.34 (m, 2H, H-2′′′, H-4′′′′), 4.21 (s, 1H, H-2′′), 4.08–3.93 (m, 8H, H-2′, H-3′′, H-3′′′, H-5′, H-5′′, H-5′′′′, H-6a′′′′, H-6b′′′′), 3.88–3.78 (m, 5H, H-2, H-3, H-3′, H-5, H-5′′′), 3.77 (s, 3H, OC6H4OCH3), 3.68 (s, 3H, OCH2C6H4OCH3), 3.59–3.53 (m, 3H, H-6a, H-6b, H-2′′′′), 3.48 (m, 1H, H-3′′′′), 2.09, 2.06, 2.01, 1.96 (4s, 12H, 4 × COCH3), 1.29 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.16 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.13 (d, 3H, J5′′′′′,6′′′′′ 6.0 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.0, 169.9, 169.7, 169.5 (4 × COCH3), 165.7, 165.6, 165.5 (3 × COC6H5), 158.9, 155.4, 151.1, 139.0, 138.5, 137.9, 137.7 (2), 137.6, 137.5, 133.0 (2), 132.8, 131.0, 130.3, 130.1, 130.0 (2), 129.8 (4), 129.2 (2), 128.4 (4), 128.3 (8), 128.2 (8), 128.1 (4), 128.1 (4), 127.8, 127.7 (2), 127.6 (2), 127.5 (4), 127.2 (4), 127.1, 118.2 (2), 114.5 (2), 113.5 (2) (ArC), 102.9 (C-1), 101.3 (C-1′′′), 100.6 (C-1′), 99.2 (C-1′′), 98.6 (C-1′′′′′), 96.6 (C-1′′′′), 81.3, 78.1, 76.1, 76.0 (2), 75.4, 75.1, 74.7, 74.6, 74.2, 73.9 (2), 73.6, 73.2 (2), 73.1, 73.0, 72.9, 72.7, 72.5, 72.2, 71.6, 71.5, 71.1, 70.4, 69.7, 69.5, 69.4, 68.5, 68.1, 67.6, 67.5, 67.5, 66.7, 64.7, 55.5, 55.0, 20.8 (2), 20.7, 20.6 (2) (4 × COCH3), 17.7, 17.6 (2), 17.4 (4 × CH3). HRMS calcd for C129H138O36Na (M + Na)+: 2285.8866, found: 2285.8863.
:1) suggested complete conversion of the starting material to a slower moving spot. The reaction mixture was washed successively with H2O and brine. Organic layer was collected, dried (Na2SO4) and evaporated in vacuo. The crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (3:1) to afford the pure compound 19 (702 mg, 78%) as foam. [α]25D +172° (c 0.7, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.11–7.11 (m, 50H, ArH), 6.99, 6.79 (2d, 4H, C6H4OCH3), 5.46–5.38 (m, 6H, H-2′′′′′, H-4, H-4′, H-4′′, H-4′′′′, H-5′′′′′), 5.28 (dd, 1H, J2′′′′′,3′′′′′ 3.5 Hz, J3′′′′′,4′′′′′ 6.5 Hz, H-3′′′′′), 5.24 (d, 1H, J1′,2′ < 1.0 Hz, H-1′), 5.16 (d, 1H, J1′′′′′,2′′′′′ < 1.0 Hz, H-1′′′′′), 5.11 (d, 1H, J1′′,2′′ < 1.0 Hz, H-1′′), 5.07 (d, 1H, J1′′′,2′′′ < 1.0 Hz, H-1′′′), 5.02–4.42 (m, 12H, 6 × CH2Ph), 4.99 (m, 1H, H-4′′′′′), 4.90 (d, 1H, J1,2 6.5 Hz, H-1), 4.43 (m, 1H, H-2′′′), 4.37 (s, 1H, H-2′′), 4.18 (t, 1H, J3′′′,4′′′, J4′′′,5′′′ 6.0 Hz, H-4′′′), 4.05–3.95 (m, 8H, H-2′, H-3′′, H-3′′′, H-5′, H-5′′, H-5′′′′, H-6a′′′′, H-6b′′′′), 3.91–3.86 (m, 3H, H-2, H-3, H-3′, H-5, H-5′′′), 3.81 (dd, 1H, J2′,3′ 7.5 Hz, J3′,4′ 2.5 Hz, H-3′), 3.77 (s, 3H, OC6H4OCH3), 3.71–3.58 (m, 3H, H-6a, H-6b, H-2′′′′), 3.47 (m, 1H, H-3′′′′), 2.06, 1.99, 1.98 (3s, 12H, 4 × COCH3), 1.29 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.21 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.17 (d, 3H, J5′′′′′,6′′′′′ 6.0 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.0, 169.9, 169.7, 169.5 (4 × COCH3), 166.0, 165.8, 165.6 (3 × COC6H5), 155.4, 151.2, 138.9, 138.5, 138.0, 137.8 (2), 137.7, 137.2, 133.3, 133.1, 132.9, 130.1, 130.0 (2), 128.5 (2), 128.4 (8), 128.3 (6), 128.2 (6). 128.1 (8), 128.0, 127.8 (2), 127.7 (2), 127.6 (2), 127.5 (2), 127.4, 127.3, 127.2, 118.3 (2), 114.5 (2) (ArC), 103.3 (C-1), 101.1 (C-1′′′), 100.6 (C-1′), 99.2 (C-1′′), 98.8 (C-1′′′′′), 95.8 (C-1′′′′), 78.2, 77.7, 76.3, 76.1, 76.0, 75.4, 75.3, 75.0, 74.9, 74.6, 73.7, 73.4, 73.2, 72.9, 72.5, 72.2, 71.7, 71.7, 71.0, 70.9, 70.8, 70.7, 70.3, 69.6, 68.9, 68.6, 68.4, 67.7, 67.6, 67.5, 67.0, 62.4, 55.6, 20.8 (2), 20.7, 20.6 (4 × COCH3), 17.8, 17.6 (2), 17.4 (4 × CH3). HRMS calcd for C121H130O35Na (M + Na)+: 2165.8290, found: 2165.8287.
:1; 20 mL). TEMPO (30 mg, 0.2 mmol) was added followed by iodosobenzene diacetate (480 mg, 1.6 mmol) and the mixture was stirred at 5 °C for 7 h. Aq. saturated Na2S2O3 (5 mL) was added to stop the reaction. After diluting the reaction mixture with CH2Cl2, it was washed with brine (2 × 30 mL). The organic layer was separated, dried (Na2SO4), filtered and evaporated to a syrupy compound. It was then dissolved in MeOH (50 mL) and passed through a 10% Pd–C cartridge in a ThalesNano flow hydrogenation assembly under continuous flow of H2 at atmospheric pressure. The hydrogenolysis of the benzyl groups were complete after 3 such cycles as evident from mass spectroscopy. NaOMe in MeOH (0.5 M, 1 mL) was added to the solution and it was stirred at room temperature for 12 h. The solution was neutralized by DOWEX 50W H+ resin, filtered and evaporated in vacuo to afford the final hexasaccharide 1 (257 mg, 75%) as white amorphous mass. [α]25D +54° (c 0.5, MeOH). HRMS calcd for C43H66O29Na (M + Na)+: 1069.3587, found: 1069.3585. 1H NMR (MeOD, 500 MHz) δ: 6.73, 6.70 (2d, 4H, C6H4OCH3), 5.26 (d, 1H, J1′,2′ ≤ 1.0 Hz, H-1′), 5.24 (d, 1H, J1′′′′′,2′′′′′ ≤ 1.0 Hz, H-1′′′′′), 5.21 (d, 1H, J1′′′′,2′′′′ ≤ 1.0 Hz, H-1′′′′), 5.11 (d, 1H, J1′′,2′′ ≤ 1.0 Hz, H-1′′), 5.06 (d, 1H, J1′′′,2′′′ ≤ 1.0 Hz, H-1′′′), 4.77 (d, 1H, J1,2 8.0 Hz, H-1), 3.70 (s, 3H, C6H4OCH3), 1.28–1.24 (m, 12H, 4 × CH3). 13C NMR (125 MHz, MeOD) δ: 173.4 (COOH), 156.5, 152.1, 116.7 (2), 115.7 (2) (aromatic C), 103.3 (C-1), 102.7 (C-1′), 102.6 (C-1′′′), 102.4 (C-1′′), 102.3 (C-1′′′′′), 101.2 (C-1′′′′), 80.0, 79.8, 78.2, 77.7, 76.6, 76.4, 74.9, 74.4, 74.2, 73.9, 73.7, 73.5, 72.3, 72.2, 72.1, 72.0, 71.9, 71.8, 70.5, 70.4, 70.3, 70.2, 69.5, 63.9, 63.2, 56.2, 18.1, 18.0, 17.9 (2) (4 × C–CH3).
:1, 36 mL) and the solution was stirred at 80 °C for 2 h until the starting material was completely converted to a slower moving spot as suggested by TLC (n-hexane–EtOAc; 3:1). The solvents were evaporated and co-evaporated twice with toluene followed by purification of the crude product by flash chromatography using (n-hexane–EtOAc; 3.5:1) to afford pure compound 3 (4.9 g, 91%) as colourless syrup. [α]25D +103° (c 1.0, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.09–7.15 (m, 9H, ArH), 5.51 (d, 1H, J1,2 1.5 Hz, H-1), 5.09 (t, 1H, J3,4, J4,5 9.5 Hz, H-4), 4.50 (m, 1H, H-5), 4.26 (bd, 1H, J1,2 1.5 Hz, H-2), 4.07 (dd, 1H, J2,3 3.0 Hz, J3,4 9.5 Hz, H-3), 3.31 (bs, 1H, OH), 2.79 (bs, 1H, OH), 2.34 (s, 3H, SC6H4CH3), 1.31 (d, 3H, J5,6 6.0 Hz). 13C NMR (CDCl3, 125 MHz) δ: 167.7 (COC6H5), 137.9, 133.6 (2), 132.1 (2), 129.9 (4), 129.3, 128.5 (2) (ArC), 87.6 (C-1), 76.6, 72.3, 70.9, 67.0, 21.1 (SC6H4CH3), 17.5 (C–CH3). HRMS calcd for C20H22O5SNa (M + Na)+: 397.1086, found: 397.1084.
:1) as eluent to give the pure compound 4 (5.0 g, 85%). [α]25D +96° (c 1.0, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.03–7.10 (m, 14H, ArH), 5.45 (d, 1H, J1,2 < 1.0 Hz, H-1), 5.42 (t, 1H, J3,4, J4,5 9.5 Hz, H-4), 4.65, 4.51 (ABq, 2H, JA–B 12.0 Hz, CH2Ph), 4.37 (m, 1H, H-5), 4.32 (dd, 1H, J1,2 < 1.0 Hz, J2,3 3.0 Hz, H-2), 3.90 (dd, 1H, J2,3 3.0 Hz, J3,4 9.5 Hz, H-3), 3.08 (bs, 1H, OH), 2.31 (s, 3H, S–C6H4–CH3), 1.24 (d, 3H, J5,6 6.5 Hz). 13C NMR (CDCl3, 125 MHz) δ: 165.9 (COC6H5), 137.9, 137.3, 133.3, 132.1 (2), 130.0 (3), 129.9 (2), 128.6 (2), 128.5 (2), 128.1 (4) (ArC), 87.4 (C-1), 77.5, 73.2, 71.7, 69.8, 67.7, 21.2 (SC6H4CH3), 17.5 (C–CH3). HRMS calcd for C27H22O5SNa (M + Na)+: 487.1555, found: 487.1553.
:1) showed complete conversion of the reactant to a faster moving spot, the mixture was evaporated in vacuo and co-evaporated twice with toluene to obtain a syrupy residue, which was then purified by flash chromatography using n-hexane–EtOAc (8:1) as eluent to give the pure compound 5 (5.2 g, 89%) as light yellow syrup. [α]25D +113° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.04–7.15 (m, 14H, ArH), 5.73 (dd, 1H, J1,2, 1.5 Hz, J2,3 3.0 Hz, H-2), 5.45 (d, 1H, J1,2 1.5, H-1), 5.36 (t, 1H, J3,4, J4,5 10.0 Hz, H-4), 4.66, 4.45 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.36 (m, 1H, H-5), 4.25, 4.17 (ABq, 2H, JA–B 15.5 Hz, COCH2Cl), 3.98 (dd, 1H, J2,3 3.0 Hz, J3,4 10.0 Hz, H-3), 2.35 (s, 3H, S–C6H4–CH3), 1.29 (d, 3H, J5,6 6.5 Hz). 13C NMR (CDCl3, 125 MHz) δ: 166.7 (COCH2Cl), 165.5 (COC6H5), 138.3, 136.8, 133.2, 132.4 (2), 129.9 (2), 129.8 (2), 129.5, 129.1, 128.3 (2), 128.2 (2), 128.0 (2), 127.8 (ArC), 86.1 (C-1), 74.2, 72.6, 71.8, 71.3, 67.9, 74.2, 72.6, 71.8, 71.3, 67.9, 40.8 (COCH2Cl), 21.0 (SC6H4CH3), 17.2 (C–CH3). HRMS calcd for C29H29ClO6SNa (M + Na)+: 563.1271, found: 563.1269.
:1) indicated complete consumption of the donor. The reaction mixture was neutralized with Et3N and the mixture was filtered through a pad of Celite. The filtrate was washed successively with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). Organic layer was separated, dried over Na2SO4 and evaporated in vacuo. The syrupy crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (4:1) as eluent to afford pure disaccharide 7 (2.3 g, 91%) as white foam. [α]25D +86° (c 0.9, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.07–7.12 (m, 20H, ArH), 7.05, 6.82 (2d, 4H, C6H4OCH3), 5.52 (dd, 1H, J2′,3′ 1.5 Hz, J1′,2′ < 1.0 Hz, H-2′), 5.39 (bd, 1H, J3,4 1.5 Hz, H-4), 5.23 (d, 1H, J1′,2′ < 1.0 Hz, H-1′), 5.22 (t, 1H, J3′,4′, J4′,5′ 10.0 Hz, H-4′), 5.06, 4.75 (ABq, 2H, JA–B 11.0 Hz, CH2Ph), 4.92 (d, 1H, J1,2 7.0 Hz, H-1), 4.61, 4.42 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.55, 4.48 (ABq, 2H, JA–B 12.0 Hz, CH2Ph), 4.21, 4.12 (ABq, 2H, JA–B 15.5 Hz, CH2Cl), 4.13 (m, 1H, H-5′), 3.93 (m, 2H, H-2, H-3), 3.88 (t, 1H, J4,5, J5,6a, J5,6b 6.5 Hz, H-5), 3.83 (dd, 1H, J2′,3′ 1.5 Hz, J3′,4′ 10.0 Hz, H-3′), 3.78 (s, 3H, OC6H4OCH3), 3.58 (m, 2H, H-6a, H-6b), 1.97 (s, 3H, COCH3), 1.25 (d, 3H, J5′,6′ 6.5 Hz). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 166.6 (COCH2Cl), 165.6 (COC6H5), 155.4, 151.1, 137.6, 137.5, 137.3, 133.1, 129.9 (2), 129.7, 128.5 (4), 128.3 (4), 128.2 (2), 127.8 (4), 127.7 (2), 127.6, 118.2 (2), 114.6 (2) (ArC), 102.9 (C-1), 98.5 (C-1′), 79.0, 75.1, 74.7, 73.6, 73.5, 72.7, 72.5, 71.1, 69.9, 69.1, 68.2, 67.3, 55.6 (OC6H4OCH3), 40.8 (COCH2Cl), 20.6 (COCH3), 17.6 (C–CH3). HRMS calcd for C51H53ClO14Na (M + Na)+: 947.3022, found: 947.3019.
:3) for 10 hours when TLC (n-hexane–EtOAc; 2.5:1) confirmed the complete conversion of the starting material to a slower moving spot. The solvents were evaporated in vacuo, the solid residue was dissolved in CH2Cl2 and washed with 1 (N) HCl (2 × 30 mL). The organic layer was collected, filtered, dried (Na2SO4) and evaporated in vacuo. The crude residue thus obtained was further subjected to flash chromatography using n-hexane–EtOAc (3:1) as eluent to give the pure disaccharide acceptor 8 (1.8 g, 87%). [α]25D +121° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.09–7.17 (m, 20H, ArH), 7.07, 6.83 (2d, 4H, C6H4OCH3), 5.42 (m, 1H, H-4), 5.34 (t, 1H, J3′,4′, J4′,5′ 9.5 Hz, H-4′), 5.30 (d, 1H, J1′,2′ < 1 Hz, H-1′), 5.04, 4.78 (ABq, 2H, JA–B 11.0 Hz, CH2Ph), 4.93 (d, 1H, J1,2 7.5 Hz, H-1), 4.61, 4.50 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.56, 4.49 (ABq, 2H, JA–B 11.5 Hz, CH2Ph), 4.10 (m, 1H, H-5′), 3.97–3.89 (m, 4H, H-2, H-3, H-5, H-2′), 3.78 (s, 3H, OC6H4OCH3), 3.73 (dd, 1H, J2′,3′ 2.0 Hz, J3′,4′ 9.5 Hz, H-3′), 3.60 (m, 2H, H-6a, H-6b), 2.60 (bs, 1H, OH), 2.03 (s, 3H, COCH3), 1.26 (d, 3H, J5′,6′ 6.5 Hz). 13C NMR (CDCl3, 125 MHz) δ: 169.8 (COCH3), 165.7 (COC6H5), 155.4, 151.3, 137.8, 137.7, 137.4, 133.0, 129.9, 129.8, 128.4 (2), 128.3 (4), 128.3 (3), 128.2 (2), 127.8, 127.7 (2), 127.7 (2), 127.6 (2), 118.3 (2), 114.5 (2) (ArC), 102.9 (C-1), 100.5 (C-1′), 79.0, 75.8, 75.4, 75.0, 73.6, 72.9, 72.8, 71.3, 69.9, 68.4, 68.2, 66.9, 55.5 (OC6H4OCH3), 20.6 (COCH3), 17.5 (C–CH3). HRMS calcd for C49H52O13Na (M + Na)+: 871.3306, found: 871.3304.
:1) confirmed the complete consumption of the donor. Then the reaction mixture was filtered through a pad of Celite. The filtrate was washed successively with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). Organic layer was separated, dried (Na2SO4) and evaporated in vacuo. The crude product thus obtained was subjected to flash chromatography using n-hexane–EtOAc (3:1) as the eluent to obtain the pure trisaccharide 9 (2.1 g, 90%) as white foam. [α]25D +87° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.14–7.12 (m, 30H, ArH), 7.01, 6.79 (2d, 4H, C6H4OCH3), 5.64 (dd, 1H, J2′′,3′′ 2.5 Hz, J1′′,2′′ < 1.0 Hz, H-2′′), 5.41 (m, 1H, H-4), 5.36 (t, 1H, J3′,4′, J4′,5′ 9.5 Hz, H-4′), 5.22 (d, 1H, J1′,2′ < 1.0 Hz, H-1′), 5.21 (t, 1H, J3′′,4′′, J4′′,5′′ 10.0 Hz, H-4′′), 4.98, 4.82 (ABq, 2H, JA–B 11.5 Hz, CH2Ph), 4.89 (m, 2H, H-1, H-1′′), 4.65–4.47 (m, 6H, 3 × CH2Ph), 4.20, 4.12 (ABq, 2H, JA–B 15.0 Hz, COCH2Cl), 4.08 (m, 1H, H-5′), 4.02 (dd, 1H, J2′′,3′′ 2.5 Hz, J3′′,4′′ 10.0 Hz, H-3′′), 3.97 (m, 1H, H-5′′), 3.89–3.86 (m, 4H, H-2, H-2′, H-3, H-5), 3.81 (dd, 1H, J2′,3′ 2.5 Hz, J3′,4′ 9.5 Hz, H-3′), 3.77 (s, 3H, OC6H4OCH3), 3.58 (m, 2H, H-6a, H-6b), 2.03 (s, 3H, COCH3), 1.30 (d, 3H, J5′,6′ 7.0 Hz, C–CH3), 1.07 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.0 (COCH3), 166.4 (COCH2Cl), 165.7 (COC6H5), 165.6 (COC6H5), 155.4, 151.2, 137.8, 137.7, 137.3, 133.1, 130.1, 129.9 (2), 129.8, 128.5 (4), 128.4 (5), 128.3 (5), 128.2 (2), 128.1 (2), 127.8 (4), 127.7, 127.6 (3), 118.3 (3), 114.6 (3) (ArC), 103.0 (C-1), 100.4 (C-1′), 99.4 (C-1′′), 78.3, 76.5, 76.0, 75.5, 74.6, 74.7, 73.5, 73.3, 72.9, 72.5, 71.0, 71.2, 70.3, 69.6, 68.4, 67.6, 67.4, 55.6 (OC6H4OCH3), 40.9 (COCH2Cl), 20.7 (COCH3), 17.8, 17.4 (2 × C–CH3). HRMS calcd for C71H73ClO19Na (M + Na)+: 1287.4332, found: 1287.4330.
:3). The mixture was refluxed for 10 h when the complete conversion of the starting material to a slower moving spot was judged by the TLC (n-hexane–EtOAc; 2:1). The mixture was evaporated in vacuo. The solid residue thus obtained was dissolved in CH2Cl2 and washed with 1 (N) HCl (2 × 30 mL) and brine (2 × 30 mL). The organic layer was separated, dried (Na2SO4) and evaporated in vacuo to get the crude product. It was purified by flash chromatography using n-hexane–EtOAc (5:2) as the eluent to get the pure compound 10 (1.7 g, 88%) as white powder. [α]25D +78° (c 0.7, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.08–7.10 (m, 30H, ArH), 6.96, 6.74 (2d, 4H, C6H4OCH3), 5.35 (m, 1H, H-4), 5.33 (t, 1H, J3′,4′, J4′,5′ 10.0 Hz, H-4′), 5.30 (t, 1H, J3′′,4′′, J4′′,5′′ 10.0 Hz, H-4′′), 5.19 (d, 1H, J1′,2′ 2.0 Hz, H-1′), 5.09 (d, 1H, J1′′,2′′ 2.0 Hz, H-1′′), 4.91, 4.82 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.86 (d, 1H, J1,2 7.5 Hz, H-1), 4.64, 4.52 (ABq, 2H, JA–B 12.0 Hz, CH2Ph), 4.56–4.42 (m, 4H, 2 × CH2Ph), 4.31 (dd, 1H, J1′′,2′′ 2.0 Hz, J2′′,3′′ 3.0 Hz, H-2′′), 4.03 (m, 1H, H-5′), 3.97 (dd, 1H, J1′,2′ 2.0 Hz, J2′,3′ 3.0 Hz, H-2′), 3.93 (m, 1H, H-5′′), 3.91 (dd, 1H, J2′′,3′′ 3.0 Hz, J3′′,4′′ 10.0 Hz, H-3′′), 3.85–3.82 (m, 3H, H-2, H-3, H-5), 3.75 (dd, 1H, J2′,3′ 3.0 Hz, J3′,4′ 10.0 Hz, H-3′), 3.69 (s, 3H, OC6H4OCH3), 3.53 (m, 2H, H-6a, H-6b), 2.62 (bs, 1H, OH), 1.93 (s, 3H, COCH3), 1.23 (d, 3H, J5′,6′ 6.5 Hz, C–CH3), 1.03 (d, 3H, J5′′,6′′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 165.7, 165.6 (2 × COC6H5), 155.3, 151.1, 137.7 (3), 137.3, 133.0, 132.9, 130.0, 129.9, 129.8 (2), 129.7 (2), 128.4 (4), 128.3 (6), 128.2 (4), 128.1 (2), 127.8, 127.7 (4), 127.6, 127.5 (2), 118.2 (2), 114.5 (2) (ArC), 103.0 (C-1), 101.0 (C-1′′), 100.6 (C-1′), 77.8, 75.9 (2), 75.6, 75.2, 74.4, 73.6, 73.2, 72.8 (2), 71.6, 71.3, 69.6, 68.4, 68.0, 67.5, 66.9, 55.5, 20.6 (COCH3), 17.7, 17.3 (2 × C–CH3). HRMS calcd for C69H72O18Na (M + Na)+: 1211.4616, found: 1211.4613.
:1) showed complete consumption of the donor. The mixture was filtered through a Celite pad and the filtrate was successively washed with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). The organic layer was collected, dried (Na2SO4) and evaporated in vacuo. Crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (2:1) as the eluent. The pure tetrasaccharide 11 (1.8 g, 89%) was obtained as white foam. [α]25D +142° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.10–7.10 (m, 40H, ArH), 6.99, 6.78 (2d, 4H, C6H4OCH3), 5.65 (dd, 1H, J1′′′,2′′′ < 1.0 Hz, J2′′′,3′′′ 3.0 Hz, H-2′′′), 5.39–5.34 (m, 3H, H-4, H-4′, H-4′′), 5.22 (d, 1H, J1′,2′ 1.5 Hz, H-1′), 5.21 (t, 1H, J3′′′,4′′′, J4′′′,5′′′ 10.0 Hz, H-4′′′), 5.14 (d, 1H, J1′′,2′′ 1.5 Hz, H-1′′), 4.96, 4.82 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.94 (d, 1H, J1′′′,2′′′ < 1.0 Hz, H-1′′′), 4.88 (d, 1H, J1,2 7.0 Hz, H-1), 4.70–4.42 (m, 8H, 4 × CH2Ph), 4.29 (dd, 1H, J1′′,2′′ 1.5 Hz, J2′′,3′′ < 1.0 Hz, H-2′′), 4.16, 4.09 (ABq, 2H, JA–B 15.5 Hz, COCH2Cl), 4.07–4.01 (m, 4H, H-3′′, H-3′′′, H-5′, H-5′′), 3.96 (m, 1H, H-5′′′), 3.95 (dd, 1H, J1′,2′ 1.5 Hz, J2′,3′ 2.5 Hz, H-2′), 3.90–3.84 (m, 3H, H-2, H-3, H-5), 3.78 (dd, 1H, J2′,3′ 2.5 Hz, J3′,4′ 9.5 Hz, H-3′), 3.76 (s, 3H, OC6H4OCH3), 3.57 (m, 2H, H-6a, H-6b), 1.97 (s, 3H, COCH3), 1.29 (d, 3H, J5′,6′ 6.5 Hz, C–CH3), 1.14 (d, 3H, J5′′,6′′ 6.5 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 166.4 (COCH2Cl), 165.8, 165.7, 165.6 (3 × COC6H5), 155.5, 151.3, 137.8, 137.7, 137.4, 133.2, 133.1 (2), 130.0 (2), 129.9 (2), 129.8 (2), 128.4 (14), 128.3 (8), 128.2 (2), 128.1 (2), 127.8 (6), 127.7 (2), 127.5 (2), 118.3 (2), 114.6 (2) (ArC), 103.1 (C-1), 101.1 (C-1′′), 100.7 (C-1′), 99.2 (C-1′′′), 78.2, 76.1, 75.7, 75.4, 74.6, 73.9, 73.7 (2), 73.3, 73.1, 72.9, 72.7, 71.8 (2), 71.3, 70.2, 69.6, 68.4, 67.7, 67.6, 67.3, 55.6, 40.9 (COCH2Cl), 20.6 (COCH3), 17.8, 17.7, 17.5 (3 × C–CH3). HRMS calcd for C91H93ClO24Na (M + Na)+: 1627.5643, found: 1627.5641.
:3), collidine (0.7 mL, 5.3 mmol) was added and the mixture was refluxed for 12 h till TLC (n-hexane–EtOAc; 2:1) indicated the complete conversion of the starting material to a slower moving spot. The reaction mixture was evaporated in vacuo and the residue was dissolved in CH2Cl2. It was further washed with 1 (N) HCl (2 × 30 mL) and brine (2 × 30 mL). Resulting organic layer was collected, dried (Na2SO4) and evaporated in vacuo to obtain the crude product. It was further purified by flash chromatography using n-hexane–EtOAc (2:1) as the eluent to obtain the pure tetrasaccharide acceptor 12 (1.5 g, 90%) as white foam. [α]25D +102° (c 0.9, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.11–7.12 (m, 40H, ArH), 6.99, 6.79 (2d, 4H, C6H4OCH3), 5.41–5.33 (m, 4H, H-4, H-4′, H-4′′, H-4′′′), 5.23 (d, 1H, J1′,2′ 1.5 Hz, H-1′), 5.19 (d, 1H, J1′′′,2′′′′ 1.5 Hz, H-1′′′), 5.12 (d, 1H, J1′′,2′′ 1.5 Hz, H-1′′), 4.95, 4.82 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.88 (d, 1H, J1,2 7.0 Hz, H-1), 4.72–4.44 (m, 8H, 4 × CH2Ph), 4.37 (dd, 1H, J1′′′,2′′′ 1.5 Hz, J2′′′,3′′′ < 1.0 Hz, H-2′′′), 4.30 (dd, 1H, J1′′,2′′ 1.5 Hz, J2′′,3′′ < 1.0 Hz, H-2′′), 4.08–4.02 (m, 4H, H-3′′, H-3′′′, H-5′, H-5′′), 4.01–3.96 (m, 2H, H-2′, H-5′′′), 3.88–3.84 (m, 3H, H-2, H-3, H-5), 3.79 (dd, 1H, J2′,3′ 3.5 Hz, J3′,4′ 6.5 Hz, H-3′), 3.77 (s, 3H, OC6H4OCH3), 3.58 (m, 2H, H-6a, H-6b), 2.57 (bs, 1H, OH), 1.96 (s, 3H, COCH3), 1.28 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.16 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.11 (d, 3H, J5′′′,6′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.4 (COCH3), 165.9, 165.8, 165.6 (3 × COC6H5), 155.4, 155.2, 127.8, 137.7 (4), 137.5, 133.1 (2), 133.0 (2), 130.6 (6), 129.8 (2), 128.4 (6), 128.3 (4), 128.2 (2), 128.0 (4), 127.8 (6), 127.7 (4), 127.6 (4), 118.3 (2), 114.5 (2) (ArC), 103.0 (C-1), 101.2 (C-1′′′), 100.9 (C-1′′), 100.6 (C-1′), 78.1, 76.4, 76.2, 76.0, 75.4 (2), 74.8, 74.6, 73.7, 73.2, 73.1, 73.0, 72.9, 71.7, 71.6, 71.5, 69.6, 68.5, 68.2, 67.6 (2), 66.9, 55.6, 20.6 (COCH3), 17.8, 17.7, 17.4 (3 × CH3). HRMS calcd for C89H92O23Na (M + Na)+: 1551.5927, found: 1551.5925.
:1) showed complete conversion of the starting material to a faster moving spot. The reaction mixture was evaporated in vacuo and co-evaporated with toluene. The crude product thus obtained was further subjected to purification by flash chromatography using n-hexane–EtOAc (6:1) as eluent to get the pure compound 14 (1.3 g, 88%). [α]25D +139° (c 1.0, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 7.54–6.96 (m, 18H, ArH), 5.74 (dd, 1H, J3,4 3.0 Hz, J4,5 < 1.0 Hz, H-4), 4.83–4.53 (m, 4H, 2 × CH2Ph), 4.67 (d, 1H, J1,2 9.0 Hz, H-1), 4.55, 4.42 (ABq, 2H, JA–B 12.5 Hz, CH2PhOMe), 4.10, 4.03 (ABq, 2H, JA–B 15.5 Hz, CH2Cl), 3.85 (s, 3H, OCH2C6H4OCH3), 3.79 (dd, 1H, J5,6a 6.5 Hz, J6a,6b 9.5 Hz, H-6a), 3.71 (dd, 1H, J2,3 9 Hz, J3,4 3 Hz, H-3), 3.65 (t, 1H, J1,2, J2,3 9.0 Hz, H-2), 3.64 (m, 1H, H-5), 3.55 (dd, 1H, J5,6b 7.5 Hz, J6a,6b 9.5 Hz, H-6b), 2.37 (s, 3H, S–C6H4–CH3). 13C NMR (CDCl3, 125 MHz) δ: 166.6 (COCH2Cl), 159.3, 138.0, 137.6, 137.3, 132.6 (2), 129.7 (2), 129.5 (2), 129.4, 129.3, 128.3 (2), 128.2 (4), 128.0 (2), 127.8, 127.7, 113.7 (2) (ArC), 87.8 (C-1), 80.9, 76.5, 75.6, 75.2, 73.1, 72.0, 68.7, 67.0, 55.1, 40.7 (COCH2Cl), 21.0 (SC6H4CH3). HRMS calcd for C37H39ClO7Na (M + Na)+: 685.2001, found: 685.1998.
:2) suggested the full consumption of the donor, the reaction was quenched by Et3N. It was then followed by filtration of the mixture through a Celite pad. Resulting solution was then successively washed with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). It was then dried (Na2SO4) and evaporated in vacuo. The crude product was then purified by flash chromatography using n-hexane–EtOAc (3:1) as the eluent. Thus the pure pentasaccharide 15 (1.4 g, 82%) was furnished as white foam. [α]25D +68° (c 0.8, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.10–6.99 (m, 55H, ArH), 6.81, 6.79 (2d, 4H, C6H4OCH3), 5.69 (bd, 1H, J3,4 1.5 Hz, H-4), 5.42–5.37 (m, 4H, H-4, H-4′, H-4′′, H-4′′′), 5.21 (d, 1H, J1′,2′ 1.0 Hz, H-1′), 5.15 (d, 1H, J1′′′,2′′′′ < 1.0 Hz, H-1′′′), 5.11 (d, 1H, J1′′,2′′ 1.0 Hz, H-1′′), 4.96, 4.83 (ABq, 2H, JA–B 12.5 Hz, CH2Ph), 4.88 (d, 1H, J1,2 7.0 Hz, H-1), 4.83, 4.76 (ABq, 2H, JA–B 10.5 Hz, CH2Ph), 4.79 (d, 1H, J1′′′′,2′′′′ 3.5 Hz, H-1′′′′), 4.70–4.15 (m, 12H, 6 × CH2Ph), 4.53 (m, 1H, H-5′′′′), 4.35 (m, 2H, H-2′′, H-2′′′), 4.12 (m, 1H, H-3′′′′), 4.08–4.01 (m, 3H, H-5′, H-3′′, H-3′′′), 4.01, 4.90 (ABq, 2H, JA–B 15.0 Hz, COCH2Cl), 3.99–3.94 (m, 3H, H-2′, H-5′′, H-5′′′), 3.87–3.86 (m, 2H, H-3, H-5), 3.81–3.78 (m, 2H, H-2, H-3′), 3.77 (s, 3H, OC6H4OCH3), 3.73 (s, 3H, OCH2C6H4OCH3), 3.64 (dd, 1H, J1′′′′,2′′′′ 3.5 Hz, J2′′′′,3′′′′ 6.5 Hz, H-2′′′′), 3.58 (m, 2H, H-6a, H-6b), 3.27 (m, 2H, H-6a′′′′, H-6b′′′′), 1.95 (s, 3H, COCH3), 1.28 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.16 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 166.7 (COCH2Cl), 165.8, 165.6 (2) (3 × COC6H5), 159.1, 155.4, 151.2, 138.8, 138.0, 137.9, 137.7 (2), 137.5, 133.1, 132.9, 130.1, 130.0, 129.9 (2), 129.8, 129.7 (2), 128.4 (10), 128.2 (10), 128.2 (10), 128.1 (4), 127.8 (4), 127.7 (2), 127.6, 127.5 (2), 127.4 (4), 127.3, 118.3 (2), 114.5 (2), 113.6 (2) (ArC), 103.0 (C-1), 101.3 (C-1′′′), 100.6 (C-1′), 99.1 (C-1′′), 97.2 (C-1′′′′), 78.1, 76.0, 75.9, 75.7, 75.5, 75.2, 75.0, 74.7, 74.6, 74.3, 73.7 (2), 73.2, 73.1, 73.0, 72.9, 72.1 (2), 71.6, 71.0, 70.3, 70.0, 68.5, 67.6 (2), 67.4, 67.2, 55.6, 55.1, 41.0 (COCH2Cl), 20.6 (COCH3), 17.8, 17.6 (2) (3 × CH3). HRMS calcd for C119H123ClO30Na (M + Na)+: 2089.7685, found: 2089.7683.
:3) was refluxed for 10 h. When the TLC (n-hexane–EtOAc; 2:1) suggested the complete conversion of the starting material to a slower moving spot, the reaction mixture was evaporated in vacuo. It was then dissolved in CH2Cl2 and washed with brine (2 × 30 mL). The organic layer was collected, dried (Na2SO4) and evaporated. The crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (2:1) to give the pure pentasaccharide acceptor 16 (1.1 g, 85%) as white foam. [α]25D +108° (c 0.7, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.07–6.98 (m, 54H, ArH), 6.78, 6.76 (2d, 4H, C6H4OCH3), 5.44–5.34 (m, 4H, H-4, H-4′, H-4′′, H-4′′′), 5.21 (d, 1H, J1′,2′ 1.0 Hz, H-1′), 5.15 (d, 1H, J1′′′,2′′′ < 1.0 Hz, H-1′′′), 5.11 (d, 1H, J1′′,2′′ 1.0 Hz, H-1′′), 4.95, 4.81 (ABq, 2H, JA–B 11.5 Hz, CH2Ph), 4.87 (d, 1H, J1,2 7.0 Hz, H-1), 4.82 (d, 1H, J1′′′′,2′′′′ 3.0 Hz, H-1′′′′), 4.70–4.40 (m, 14H, 7 × CH2Ph), 4.37–4.36 (m, 3H, H-2′′, H-2′′′, H-4′′′′), 4.13 (m, 1H, H-5′′′′), 4.06–3.90 (m, 8H, H-2′, H-3′′, H-3′′′, H-5′, H-5′′, H-5′′′, H-6a′′′′, H-6b′′′′), 3.80–3.79 (m, 4H, H-2, H-3, H-3′, H-5), 3.77 (s, 3H, OC6H4OCH3), 3.70 (s, 3H, OCH2C6H4OCH3), 3.58 (m, 3H, H-6a, H-6b, H-2′′′′), 3.64 (m, 1H, H-3′′′′), 2.82 (br s, 1H, OH), 1.94 (s, 3H, COCH3), 1.27 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.16 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 169.9 (COCH3), 165.7 (2), 165.6 (3 × COC6H5), 159.1, 155.5, 151.2, 139.0, 138.0, 137.8 (2), 137.7, 137.6, 133.1, 132.8, 130.2, 130.1 (2), 129.9 (2), 129.8 (4), 129.4 (2), 128.4 (10), 128.3 (4), 128.2 (8), 128.1 (6), 127.9 (4), 127.8 (4), 127.7 (2), 127.6 (2), 127.5 (2), 127.3 (2), 118.3 (2), 114.6 (2), 113.7 (2) (ArC), 103.0 (C-1), 101.3 (C-1′′′), 100.7 (C-1′), 99.2 (C-1′′), 96.8 (C-1′′′′), 78.1, 76.0, 75.5, 74.6, 73.7 (2), 73.3 (4), 73.2 (2), 73.0, 72.7, 72.4, 72.2, 71.6, 70.8, 69.6, 69.4, 68.5, 68.3, 68.2 (2), 67.7, 67.6 (2), 55.6, 55.1, 20.6 (COCH3), 17.8, 17.7, 17.6 (3 × CH3). HRMS calcd for C117H122O29Na (M + Na)+: 2013.7969, found: 2013.7967.
:1) suggested that the whole of the acceptor was consumed, the mixture was neutralised with Et3N and filtered through a Celite pad. The filtrate was successively washed with aq. Na2S2O3 (2 × 30 mL), aq. NaHCO3 (2 × 30 mL) and brine (30 mL). The organic layer was separated, dried (Na2SO4) and evaporated in vacuo to get the crude product. It was purified by flash chromatography using n-hexane–EtOAc (3:2) as eluent to obtain the pure hexasaccharide 18 (960 mg, 84%) as the white foam. [α]25D +132° (c 0.7, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.11–7.00 (m, 54H, ArH), 6.99, 6.79 (2d, 4H, C6H4OCH3), 5.50 (s, 1H, H-2′′′′′), 5.44–5.39 (m, 4H, H-4, H-4′, H-4′′, H-4′′′), 5.37–5.34 (m, 2H, H-3′′′′′, H-5′′′′′), 5.21 (m, 2H, H-1′, H-1′′′′′), 5.09 (m, 2H, H-1′′, H-1′′′), 5.03 (t, 1H, J3′′′′′,4′′′′′, J4′′′′′,5′′′′′ 10.0 Hz, H-4′′′′′), 4.96, 4.83 (ABq, 2H, JA–B 11.5 Hz, CH2Ph), 4.89 (d, 1H, J1,2 6.5 Hz, H-1), 4.78–4.28 (m, 14H, 7 × CH2Ph), 4.70 (d, 1H, J1′′′′,2′′′′ 3.0 Hz, H-1′′′′), 4.36–4.34 (m, 2H, H-2′′′, H-4′′′′), 4.21 (s, 1H, H-2′′), 4.08–3.93 (m, 8H, H-2′, H-3′′, H-3′′′, H-5′, H-5′′, H-5′′′′, H-6a′′′′, H-6b′′′′), 3.88–3.78 (m, 5H, H-2, H-3, H-3′, H-5, H-5′′′), 3.77 (s, 3H, OC6H4OCH3), 3.68 (s, 3H, OCH2C6H4OCH3), 3.59–3.53 (m, 3H, H-6a, H-6b, H-2′′′′), 3.48 (m, 1H, H-3′′′′), 2.09, 2.06, 2.01, 1.96 (4s, 12H, 4 × COCH3), 1.29 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.16 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.13 (d, 3H, J5′′′′′,6′′′′′ 6.0 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.0 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.0, 169.9, 169.7, 169.5 (4 × COCH3), 165.7, 165.6, 165.5 (3 × COC6H5), 158.9, 155.4, 151.1, 139.0, 138.5, 137.9, 137.7 (2), 137.6, 137.5, 133.0 (2), 132.8, 131.0, 130.3, 130.1, 130.0 (2), 129.8 (4), 129.2 (2), 128.4 (4), 128.3 (8), 128.2 (8), 128.1 (4), 128.1 (4), 127.8, 127.7 (2), 127.6 (2), 127.5 (4), 127.2 (4), 127.1, 118.2 (2), 114.5 (2), 113.5 (2) (ArC), 102.9 (C-1), 101.3 (C-1′′′), 100.6 (C-1′), 99.2 (C-1′′), 98.6 (C-1′′′′′), 96.6 (C-1′′′′), 81.3, 78.1, 76.1, 76.0 (2), 75.4, 75.1, 74.7, 74.6, 74.2, 73.9 (2), 73.6, 73.2 (2), 73.1, 73.0, 72.9, 72.7, 72.5, 72.2, 71.6, 71.5, 71.1, 70.4, 69.7, 69.5, 69.4, 68.5, 68.1, 67.6, 67.5, 67.5, 66.7, 64.7, 55.5, 55.0, 20.8 (2), 20.7, 20.6 (2) (4 × COCH3), 17.7, 17.6 (2), 17.4 (4 × CH3). HRMS calcd for C129H138O36Na (M + Na)+: 2285.8866, found: 2285.8863.
:1) suggested complete conversion of the starting material to a slower moving spot. The reaction mixture was washed successively with H2O and brine. Organic layer was collected, dried (Na2SO4) and evaporated in vacuo. The crude product thus obtained was purified by flash chromatography using n-hexane–EtOAc (3:1) to afford the pure compound 19 (702 mg, 78%) as foam. [α]25D +172° (c 0.7, CHCl3). 1H NMR (CDCl3, 500 MHz) δ: 8.11–7.11 (m, 50H, ArH), 6.99, 6.79 (2d, 4H, C6H4OCH3), 5.46–5.38 (m, 6H, H-2′′′′′, H-4, H-4′, H-4′′, H-4′′′′, H-5′′′′′), 5.28 (dd, 1H, J2′′′′′,3′′′′′ 3.5 Hz, J3′′′′′,4′′′′′ 6.5 Hz, H-3′′′′′), 5.24 (d, 1H, J1′,2′ < 1.0 Hz, H-1′), 5.16 (d, 1H, J1′′′′′,2′′′′′ < 1.0 Hz, H-1′′′′′), 5.11 (d, 1H, J1′′,2′′ < 1.0 Hz, H-1′′), 5.07 (d, 1H, J1′′′,2′′′ < 1.0 Hz, H-1′′′), 5.02–4.42 (m, 12H, 6 × CH2Ph), 4.99 (m, 1H, H-4′′′′′), 4.90 (d, 1H, J1,2 6.5 Hz, H-1), 4.43 (m, 1H, H-2′′′), 4.37 (s, 1H, H-2′′), 4.18 (t, 1H, J3′′′,4′′′, J4′′′,5′′′ 6.0 Hz, H-4′′′), 4.05–3.95 (m, 8H, H-2′, H-3′′, H-3′′′, H-5′, H-5′′, H-5′′′′, H-6a′′′′, H-6b′′′′), 3.91–3.86 (m, 3H, H-2, H-3, H-3′, H-5, H-5′′′), 3.81 (dd, 1H, J2′,3′ 7.5 Hz, J3′,4′ 2.5 Hz, H-3′), 3.77 (s, 3H, OC6H4OCH3), 3.71–3.58 (m, 3H, H-6a, H-6b, H-2′′′′), 3.47 (m, 1H, H-3′′′′), 2.06, 1.99, 1.98 (3s, 12H, 4 × COCH3), 1.29 (d, 3H, J5′,6′ 6.0 Hz, C–CH3), 1.21 (d, 3H, J5′′,6′′ 6.0 Hz, C–CH3), 1.17 (d, 3H, J5′′′′′,6′′′′′ 6.0 Hz, C–CH3), 1.09 (d, 3H, J5′′′,6′′′ 6.5 Hz, C–CH3). 13C NMR (CDCl3, 125 MHz) δ: 170.0, 169.9, 169.7, 169.5 (4 × COCH3), 166.0, 165.8, 165.6 (3 × COC6H5), 155.4, 151.2, 138.9, 138.5, 138.0, 137.8 (2), 137.7, 137.2, 133.3, 133.1, 132.9, 130.1, 130.0 (2), 128.5 (2), 128.4 (8), 128.3 (6), 128.2 (6). 128.1 (8), 128.0, 127.8 (2), 127.7 (2), 127.6 (2), 127.5 (2), 127.4, 127.3, 127.2, 118.3 (2), 114.5 (2) (ArC), 103.3 (C-1), 101.1 (C-1′′′), 100.6 (C-1′), 99.2 (C-1′′), 98.8 (C-1′′′′′), 95.8 (C-1′′′′), 78.2, 77.7, 76.3, 76.1, 76.0, 75.4, 75.3, 75.0, 74.9, 74.6, 73.7, 73.4, 73.2, 72.9, 72.5, 72.2, 71.7, 71.7, 71.0, 70.9, 70.8, 70.7, 70.3, 69.6, 68.9, 68.6, 68.4, 67.7, 67.6, 67.5, 67.0, 62.4, 55.6, 20.8 (2), 20.7, 20.6 (4 × COCH3), 17.8, 17.6 (2), 17.4 (4 × CH3). HRMS calcd for C121H130O35Na (M + Na)+: 2165.8290, found: 2165.8287.
:1; 20 mL). TEMPO (30 mg, 0.2 mmol) was added followed by iodosobenzene diacetate (480 mg, 1.6 mmol) and the mixture was stirred at 5 °C for 7 h. Aq. saturated Na2S2O3 (5 mL) was added to stop the reaction. After diluting the reaction mixture with CH2Cl2, it was washed with brine (2 × 30 mL). The organic layer was separated, dried (Na2SO4), filtered and evaporated to a syrupy compound. It was then dissolved in MeOH (50 mL) and passed through a 10% Pd–C cartridge in a ThalesNano flow hydrogenation assembly under continuous flow of H2 at atmospheric pressure. The hydrogenolysis of the benzyl groups were complete after 3 such cycles as evident from mass spectroscopy. NaOMe in MeOH (0.5 M, 1 mL) was added to the solution and it was stirred at room temperature for 12 h. The solution was neutralized by DOWEX 50W H+ resin, filtered and evaporated in vacuo to afford the final hexasaccharide 1 (257 mg, 75%) as white amorphous mass. [α]25D +54° (c 0.5, MeOH). HRMS calcd for C43H66O29Na (M + Na)+: 1069.3587, found: 1069.3585. 1H NMR (MeOD, 500 MHz) δ: 6.73, 6.70 (2d, 4H, C6H4OCH3), 5.26 (d, 1H, J1′,2′ ≤ 1.0 Hz, H-1′), 5.24 (d, 1H, J1′′′′′,2′′′′′ ≤ 1.0 Hz, H-1′′′′′), 5.21 (d, 1H, J1′′′′,2′′′′ ≤ 1.0 Hz, H-1′′′′), 5.11 (d, 1H, J1′′,2′′ ≤ 1.0 Hz, H-1′′), 5.06 (d, 1H, J1′′′,2′′′ ≤ 1.0 Hz, H-1′′′), 4.77 (d, 1H, J1,2 8.0 Hz, H-1), 3.70 (s, 3H, C6H4OCH3), 1.28–1.24 (m, 12H, 4 × CH3). 13C NMR (125 MHz, MeOD) δ: 173.4 (COOH), 156.5, 152.1, 116.7 (2), 115.7 (2) (aromatic C), 103.3 (C-1), 102.7 (C-1′), 102.6 (C-1′′′), 102.4 (C-1′′), 102.3 (C-1′′′′′), 101.2 (C-1′′′′), 80.0, 79.8, 78.2, 77.7, 76.6, 76.4, 74.9, 74.4, 74.2, 73.9, 73.7, 73.5, 72.3, 72.2, 72.1, 72.0, 71.9, 71.8, 70.5, 70.4, 70.3, 70.2, 69.5, 63.9, 63.2, 56.2, 18.1, 18.0, 17.9 (2) (4 × C–CH3).
Acknowledgements
VS is thankful to IISER Kolkata for Senior Research Fellowship. SERB, DST, India is gratefully acknowledged for funding through grant SB/S1/OC-48/2013 to BM.Notes and references
- R. Roy, Drug Discovery Today: Technol., 2004, 1, 327–336 CrossRef CAS PubMed.
- K. J. Doores, D. P. Gamblin and B. G. Davis, Chem.–Eur. J., 2006, 12, 656–665 CrossRef CAS PubMed.
- B. Kuberan and R. J. Linhardt, Curr. Org. Chem., 2000, 4, 653–677 CrossRef CAS.
- R. Podschun and U. Ullmann, Clin. Microbiol. Rev., 1998, 11, 589–603 CAS.
- J. B. Robbins, R. Schneerson, S. C. Szu, A. Fattom, Y. Yang, T. Lagergard, C. Chu and U. S. Sorensen, Curr. Top. Microbiol. Immunol., 1989, 146, 169–180 CAS.
- S. J. Cryz Jr, P. Mortimer, A. S. Cross, E. Furer and R. Germanier, Vaccine, 1986, 4, 15–20 CrossRef CAS.
- J. Kubler-Kielb, E. Vinogradov, W.-I. Ng, B. Maczynska, A. Junka, M. Bartoszewicz, A. Zelazny, J. Bennett and R. Schneerson, Carbohydr. Res., 2013, 369, 6–9 CrossRef CAS PubMed.
- B. Mukhopadhyay, K. P. R. Kartha, D. A. Russell and R. A. Field, J. Org. Chem., 2004, 69, 7758–7760 CrossRef CAS PubMed.
- P. A. Gent and R. Gigg, J. Chem. Soc., Perkin Trans. 1, 1974, 1446–1455 RSC.
- V. K. Rajput and B. Mukhopadhyay, J. Org. Chem., 2008, 73, 6924–6927 CrossRef CAS PubMed.
- M. J. Bertolini and C. P. J. Glaudemans, Carbohydr. Res., 1970, 15, 263–270 CrossRef CAS.
- V. Sarkar and B. Mukhopadhyay, Carbohydr. Res., 2015, 406, 65–70 CrossRef CAS PubMed.
- (a) S. Dasgupta, B. Roy and B. Mukhopadhyay, Carbohydr. Res., 2006, 341, 2708–2713 CrossRef CAS PubMed; (b) K. B. Pal and B. Mukhopadhyay, Carbohydr. Res., 2013, 379, 26–29 CrossRef CAS PubMed; (c) P. R. Verma and B. Mukhopadhyay, RSC Adv., 2013, 3, 201–207 RSC; (d) D. Budhadev and B. Mukhopadhyay, RSC Adv., 2015, 5, 98033–98040 RSC.
- K. Takeo, K. Maki, Y. Wada and S. Kitamura, Carbohydr. Res., 1993, 245, 81–96 CrossRef CAS PubMed.
- D. Budhadev and B. Mukhopadhyay, Tetrahedron, 2015, 71, 6155–6163 CrossRef CAS.
- I. Cumpstey, A. J. Fairbanks and A. J. Redgrave, Tetrahedron, 2004, 60, 9061–9074 CrossRef CAS.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 885–888 CrossRef CAS.
- L. J. Van den Bos, J. D. C. Codee, J. C. van der Toorn, T. J. Boltje, J. H. van Boom, H. S. Overkleeft and G. A. van der Marel, Org. Lett., 2004, 6, 2165–2168 CrossRef CAS PubMed.
- G. Zemplen, Ber. Dtsch. Chem. Ges., 1926, 59, 1254–1266 CrossRef.
- D. D. Perrin, W. L. Amarego and D. R. Perrin, Purification of laboratory chemicals, Pergamon, London, 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of all new compounds. See DOI: 10.1039/c6ra07351d |
| This journal is © The Royal Society of Chemistry 2016 |