
A gadolinium MOF acting as a multi-responsive and highly selective luminescent sensor for detecting o-, m-, and p-nitrophenol and Fe3+ ions in the aqueous phase†
Abstract
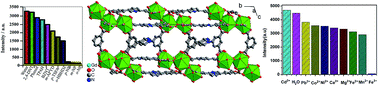
A microporous Gd-MOF, [Gd6(L)3(HL)2(H2O)10]·18H2O·x(solvent) (1), has been successfully synthesized by a solvothermal reaction between Gd(NO3)3·6H2O and the multidentate π-conjugated ligand, H4L, which has a Lewis basic pyridyl site (H4L = 5,5′-(pyridine-2,5-diyl)-isophthalic acid). The crystal structure shows that compound 1 consists of Gd3 units, which are further interlinked by multicarboxylate ligands to form a 2D network. A solid sample of 1 emits bright blue light, which can be assigned to H4L ligand-centered emission. Interestingly, the luminescence of finely ground particles of 1 dispersed in water shows high sensitivity and selectivity towards trace amounts of o-, m-, and p-nitrophenol (NP) and Fe3+ ions with good linearity, which indicates that 1 can be used as a multi-responsive luminescence sensor for the detection of o-, m-, and p-NP and Fe3+ ions in an aqueous system.
- This article is part of the themed collection: RSC Advances Editors' collection: f Block Chemistry
 Please wait while we load your content...
Please wait while we load your content...

