
A study of solvent selectivity on the crystal morphology of FOX-7 via a modified attachment energy model
Qiangli
Zhao
a,
Ning
Liu
b,
Bozhou
Wang
*b and
Wenliang
Wang
*a
aKey Laboratory for Macromolecular Science of Shaanxi Province, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710062, Shaanxi, China. E-mail: wlwang@snnu.edu.cn; wbz600@163.com; Fax: +86-29-81530727; Tel: +86-29-81530815
bXi’an Modern Chemistry Research Institute, Xi’an 710065, Shaanxi, China
First published on 15th June 2016
Abstract
The crystal morphology of FOX-7 in different solvents was investigated via molecular dynamics simulations. A modified attachment energy (MEA) model was constructed by introducing surface chemistry terms and the associated topography of the habit crystal plane. Solvent has a large effect on the crystal morphology of FOX-7. The calculated results show that the crystal morphology of FOX-7 in vacuum is dominated by six faces, (0 1 1), (1 0 −1), (1 0 1), (1 1 −1), (0 0 2) and (1 10), while that in acetic acid is dominated by three faces, (1 1 −1), (0 1 1) and (1 0 1). The applicability of the model was successfully validated by the study of FOX-7 crystals grown from cyclohexanone, acetonitrile, H2O/DMF, and H2O/DMSO. All the predicted results are in good agreement with the experimental results. The crystal morphology of FOX-7 is variable in different crystallization solvents. H2O/DMF and H2O/DMSO are good choices for the solvent recrystallization of FOX-7. The results are useful for the formulation design of FOX-7.
1. Introduction
1,1-Diamino-2,2-dinitroethene (FOX-7, C2H4N4O4) has been referred to as a promising candidate for secondary explosives because of its excellent explosive performance and quite low sensitivity. Due to these attractive characteristics, many studies have been performed on its synthesis,1,2 polymorphism,3–7 thermal analysis,8–11 and crystallization.12–16 Of particular interest is the control of crystal morphology during crystallization steps, as crystal morphology has a great impact on the performance and shock sensitivity of energetic materials17 as well as the efficiency of filtering and washing during downstream processes. Compared with needle-shaped crystals, spherical crystals can improve the packing density and impact sensitivity largely. Therefore, research into the control of FOX-7 crystal morphology is of important practical significance.Crystal morphology is governed by two factors, the crystal internal structure and external conditions such as crystallization technology,13,18,19 and the presence of solvent20–22 and impurities.23,24 Among these, solvent is believed to be one of the most important factors, which dramatically changes the relative growth rate and consequently the crystal habit. To date, progress in understanding crystallization in energetic crystals has been achieved through a concerted experimental25,26 and computational approaches.27,28 Moreover, computational simulations can provide enough microscopic details of crystallization, and have proven to be a powerful and important tool in studying crystallization processes on the atomistic/molecular length scale in recent years. Duan et al.29 investigated the solvent effect on octahydro-1,3,5,7-tetranititro-1,3,5,7-tetrazocine (HMX) in acetone by molecular dynamics simulations, and they modified the attachment energy model by incorporating the solvent effect on crystal morphology as a corrected term for attachment energy in vacuum. Shi et al.30 investigated the effect of trifluoroacetic acid on the crystal morphology of 2,6-diamino-3,5-dinitropyridine-1-oxide (ANPyO) using molecular dynamic simulations. They found that the growth habit was also affected by the diffusion capacity of solvent molecules. Chen et al.31,32 discussed the crystal habits of hexogen in acetone and cyclotrimethylene trinitramine in cyclohexanone (CYC) via dynamic simulations based on 2D nucleation theory. They successfully predicted the crystal morphologies. Lee et al.33 attempted to explain the co-solvent effect on the shape evolution of 7-amino-4,6-dinitrobenzofuroxan (ADNBF) crystals using a molecular modelling technique. They obtained the crystal morphology of ADNBF grown from co-solvents by the modified attachment energy model with a term for the binding site densities of crystal surfaces. Zhang et al.34 proposed a new occupancy model for predicting crystal morphology influenced by solvent and temperature. The model was validated by successfully predicting the crystal morphology of some nitroamino explosives. Research into the growth morphology of FOX-7 at the molecular level has made some progress. Shim et al.35 explored the cooling crystallization process by molecular modelling. Through step energy calculations and kinetic Monte Carlo simulations, the supersaturation-dependent growth habit properties of FOX-7 were analyzed. Qian et al.36 performed simulations to study the anisotropy of thermal expansion for crystalline FOX-7. Ren et al.37 analyzed the crystal morphology and crystallization behavior of FOX-7 in DMF/acetone solvent by molecular dynamics simulation. Although previous work has achieved many important and meaningful results, the relationship between solvent effect and FOX-7 crystal habit is still unclear and requires further clarification.
Pursuing our interests in exploring the effect of solvent on the crystal morphology of FOX-7, and further obtaining solvent selectivity for the crystal morphology of FOX-7, we initiated work to investigate the crystal morphology of FOX-7 grown from different solvents by molecular dynamics simulations. We carried out molecular dynamics simulations to predict the crystal morphology of FOX-7 grown from acetic acid, cyclohexanone, acetonitrile, water/N,N-dimethyl formamide (H2O/DMF), and water/dimethyl sulfoxide (H2O/DMSO). We focused on the solvent and co-solvent effects on the crystal morphology arising from different solvents. In Section 2, the theoretical model and the simulation details are described. Section 3 gives the experimental details. Section 4 presents the experimental and calculated results for the equilibrium crystal morphology of FOX-7 crystals in different solvents. By the attachment energy model, FOX-7 growth habits in vacuum are determined. Taking the system of FOX-7 in acetic acid solvent as an example, the solvent effect on crystal morphology is investigated. By analyzing the attachment energies, relative growth energy, as well as the growth habit in different solvents, solvent selectivity for the crystal morphology is obtained. The calculated results are validated by the experimental results. Section 5 summarizes the final conclusions. The predicted morphologies would help us to understand the solvent selectivity of the FOX-7 crystal, which would provide a guide for high-quality FOX-7 preparation.
2. Theoretical model and computational simulations
2.1 Theoretical model
Here, the attachment energy (AE) model, which takes into account the anisotropic energies in the crystal unit cell and attempts to simulate crystal habits as obtained under non-equilibrium growth conditions, was chosen to determine morphologically possible growth faces. The model, which was proposed by Hartman and Bennema based upon period bond chain (PBC) theory,38 relates the relative growth rate of a given surface to the potential energy per unit cell gained if a new layer of material attaches to the surface in vacuum. The attachment energy, Eatt, is defined as the energy released on attachment of a growth slice to a growing crystal surface.39 Eatt is calculated as| Eatt = Elatt − Ea | (1) |
| Rhkl ∝ Eatthkl | (2) |
The AE model is usually applied to the prediction of crystal morphology in the vapor phase. However, due to its disregard for the external crystallization conditions, it fails to predict crystal habits grown from solution. As mentioned above, the crystal growth from solution is determined by not only the internal crystal structures but also the external growth conditions, such as supersaturation, solvent, etc. These factors have great influence on the crystal morphology. Obviously, for the better prediction of crystal habit in solution, the AE model should be modified to consider the growth environment. The growth of FOX-7 crystals is regarded as an epitaxial process of a growth interface and the growth interface determines the crystal growth mechanism. Based on the two-dimensional nucleation theory40 and the relationship between the attachment energy of the crystal face and its surface free energy, the growth rate of FOX-7 face in solution can be expressed as:
| Rsolhkl ∝ Eatt,mhkl | (3) |
 | (4) |
2.2 Computational simulations
All computational simulations were performed using Materials Studio 5.5 software.41 FOX-7 is described as a push–pull ethylene with two electron donating amino groups (“heads”) on one carbon and two electron withdrawing nitro groups (“tails”) on the other carbon within its molecule framework. The molecules in the crystal are arranged as wave-shaped layers with extensive intermolecular hydrogen bonding in the layers and weak van der Waals (vdW) forces between the layers. The crystal structure of FOX-7 (ref. 42) with space group P21/n, Z = 4, a = 6.94 Å, b = 6.64 Å, c = 11.34 Å, and β = 90.6° is displayed in Fig. 1. | ||
| Fig. 1 The crystal structure of FOX-7. | ||
The built-in ab initio COMPASS force field43 was employed to the geometry optimizations and MD simulations. The COMPASS force field enables the accurate and simultaneous prediction of structural, conformational, vibrational, and thermophysical properties for a broad range of molecules in isolation or condensed phases under a wide range of temperature and pressure conditions.44 Through the geometrical optimization of a unit cell (a = 6.20 Å, b = 6.51 Å, c = 12.33 Å and β = 92.3°), the calculated lattice energy was −25.62 kcal mol−1, which agrees well with an experimental lattice energy of −27.08 kcal mol−1.45 By optimizing the 3*3*3 crystal supercell, an optimized cell with a = 6.50 Å, b = 6.94 Å, c = 11.04 Å and β = 92.2° was obtained.
The AE model was selected to predict the crystal morphology of FOX-7 in vacuum, acquiring a list of morphologically possible growth faces. Then the FOX-7 crystal was cleaved parallel to the predicted (h k l) faces at a depth of three unit cells, and each crystal slice was constructed as a periodic superstructure of a 3*3 unit cell. By using the Amorphous Cell tool, the three-dimensional periodic solvent box with 100 randomly distributed solvent molecules was constructed and then refined by MD techniques. The dimensions of the solvent box were consistent with the lattice parameters of the selected crystal surface. Geometry optimization, followed by MD simulations (1000 ps with a time step of 1 fs at 298 K, using the Andersen thermostat ensemble46) for the solvent layer was done to make solvent molecules uniformly distributed in the solvent layer. The FOX-7 surface–solvent micro interface model was constructed to study solvent effect on crystal morphology. A vacuum slab with a thickness of 30 Å was built above the solvent layer to eliminate the effect of additional free boundaries on the structure. The isolated FOX-7 surface layer was obtained by deleting the solvent layer. When deleting the surface layer, the isolated solvent layer was achieved. All geometry optimizations and MD simulations were performed in the COMPASS force field. MD simulations with an NVT ensemble were performed at 298 K controlled by an Andersen thermostat after geometry optimization of the initial interface models. To achieve system equilibrium, a period of 1000 ps dynamics simulations with a time step of 1.0 fs was performed. MD simulations with a period of 500 ps were carried out for the production stage, during which the dynamic trajectories were collected every 100 time steps. For the potential energy calculations, the calculations of van der Waals interactions and Coulomb interactions were processed through an atom based method with a cutoff distance of 12.5 Å and the Ewald summation method, respectively. The adsorption energy, Eshkl, values were averaged from the final 500 frames of the dynamic trajectory (last 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps), calculated using the following equation:
000 steps), calculated using the following equation:
 | (5) |
3. Experimental
3.1 Materials and characterization
FOX-7 material was prepared according to the literature.52 It had a mass fraction purity of 99%. Acetic acid (AR), cyclohexanone (AR), DMF (AR), DMSO (AR) and deionized water were used. The images of the crystal morphologies were obtained using a scanning electron microscope system (Quanta 600 FEG).3.2 Recrystallization
50 mL FOX-7 solutions were prepared with 0.5 g FOX-7 added into acetic acid, cyclohexanone, acetonitrile, and co-solvent (DMSO:H2O = 1:1 in volume ratio) or (DMF:H2O = 1:1 in volume ratio), respectively. The solution was heated from room temperature to 90 °C by stirring at a certain speed in a water bath to make it completely dissolved. Afterwards, the resulting solution was naturally cooled down to room temperature and seed crystals were obtained with spontaneous nucleation.
4. Results and discussion
Here we mainly focus on the solvent effect on crystal habit. The crystal morphologies are governed by the crystal internal structures, the adsorption interaction, and the surface structure. The effects from the internal structures can be analyzed by the AE model. The contributions from the latter two factors can be characterized by the FOX-7 surface–solvent micro interface model and a parameter S defined as the ratio of the solvent-accessible area to the corresponding crystal face area.4.1 Crystal morphology of FOX-7 in vacuum
Fig. 2 shows the crystal morphology of FOX-7 in vacuum calculated by the AE model using force field assigned charges and the COMPASS force field, and Table 1 lists the relevant parameters of the main crystal habit faces of FOX-7. | ||
| Fig. 2 (a) Crystal morphology and (b) crystal graph of FOX-7 in vacuum predicted by the AE model. Different coloured lines represent different bonds. The numbers represent the molecular interactions, energies in kcal mol−1. | ||
| (h k l) | Multiplicity | d hkl | E att | Distance | Total facet area/% |
|---|---|---|---|---|---|
| a All energies are in kcal mol−1, distance is in Å. | |||||
| (0 1 1) | 4 | 5.73 | −30.63 | 30.63 | 42.86 |
| (1 0 −1) | 2 | 5.95 | −28.03 | 28.03 | 24.31 |
| (1 0 1) | 2 | 5.90 | −37.15 | 37.15 | 15.97 |
| (1 1 −1) | 4 | 4.43 | −34.51 | 34.51 | 9.13 |
| (0 0 2) | 2 | 5.67 | −43.60 | 43.60 | 4.90 |
| (1 1 0) | 4 | 4.80 | −39.91 | 39.91 | 2.83 |
From Fig. 2a, it can be seen that the prediction of crystal shape according to the AE model results in a shape similar to the fusiform. The ratio between the longest and the shortest diameter of the habit is 1.702. The molecular interactions between FOX-7 molecules were calculated by morphology and visualized by crystal graph as shown in Fig. 2b. It has been proven that the growth velocity of crystal increases as the interaction becomes stronger. As a result, the morphology of FOX-7 crystal in vacuum is dominated by six faces, (0 1 1), (1 0 −1), (1 0 1), (1 1 −1), (0 0 2) and (1 1 0). Table 1 shows that the (0 1 1), (1 0 −1) and (1 0 1) planes are the most important faces, of which the (0 1 1) face has the largest habit face area. There is a tendency that (1 1 −1), (0 0 2) and (1 1 0) are more likely to disappear. Based on the important faces calculated using the AE model, the morphology of FOX-7 in acetic acid, cyclohexanone, acetonitrile, H2O/DMF, and H2O/DMSO was predicted.
4.2 Crystal morphology of FOX-7 in solvent
The equilibrium configurations of FOX-7 surface–acetic acid solvent adsorption interface models are displayed in Fig. 3. From these snapshots, it can be seen that acetic acid molecules closely attach to FOX-7 surfaces and form acetic acid layers, which implies that there are strong adsorption interactions between FOX-7 surfaces and acetic acid solvent. Moreover, the distribution of acetic acid is mainly concentrated on the solvent accessible surface of FOX-7 crystal faces as shown in Fig. 3. That is to say that, the morphology change influenced by the solvent is a result of a combination of the adsorption interaction and the surface structure. | ||
| Fig. 3 Snapshots of equilibrium configurations of different FOX-7 surface–acetic acid solvent adsorption models. Blue is the crystal, black is the adsorbing molecules and grey is the solvent. | ||
The adsorption energies (Eshkl) of acetic acid solvent adsorbed on different FOX-7 surfaces are listed in Table 2. As seen in Table 2, it is found that the (1 0 1) face has the maximum Eshkl of −246.88 kcal mol−1 (the minus sign represents an exothermic process), while the (1 1 0) face has the minimum Eshkl of 50.57 kcal mol−1. The interaction strength of acetic acid with different crystal faces can be compared in the following sequence (1 0 1) > (1 1 −1) > (0 1 1) > (0 0 2) > (1 0 −1) > (1 1 0). The adsorption energy reflects the affinity capacity of solvents on the particular surfaces. Therefore, the adsorption ability of acetic acid solvent with the (1 0 1) face is the strongest while the adsorption interaction between acetic acid molecules and the (1 1 0) face is the weakest. It is supposed that the surface diffusion solvent molecules would adsorb on the surface, which can block the incorporation of solute molecules on the bonding sites and result in the inhibition of crystal surface growth. Hence, solvent–surface adsorption interaction is usually considered as an important factor in changing crystal habit and can represent the solvent effect to a large extent.
| (h k l) | E tot,avg hkl | E sur,avg hkl | E sol,avg hkl | E s hkl |
|---|---|---|---|---|
| a All energies are in kcal mol−1. b The standard deviations used as the estimated errors. | ||||
| (1 0 −1) | −31898.35 | −27524.44 | −4238.06 | −135.85(±11.79)b |
| (0 1 1) | −31788.44 | −27327.28 | −4254.55 | −206.62(±13.60) |
| (1 1 −1) | −31645.87 | −27211.96 | −4222.28 | −211.63(±13.93) |
| (1 0 1) | −31403.23 | −27413.91 | −4242.55 | −246.88(±14.19) |
| (1 1 0) | −31403.23 | −27224.53 | −4229.27 | 50.57(±13.23) |
| (0 0 2) | −18042.24 | −13566.50 | −4325.22 | −150.52(±13.41) |
Fig. 4 shows the molecular packing structures of FOX-7. It shows that the molecular arrangement of the (1 0 −1), (0 1 1) and (1 0 1) planes are relatively flat at the molecular level, while other surfaces are uneven with many large voids, which may contribute to solvent molecule adsorption. The S parameter values of FOX-7 surfaces calculated by the Connolly surface model are listed in Table 3. This table shows that among the FOX-7 habit surfaces, the (1 1 −1) face has the maximum S value of 1.25, which indicates larger morphological roughness. In contrast, the (1 0 −1) face has the minimum, which indicates larger morphological smoothness. A larger S value indicates a more complex surface topography, which is thus more convenient for solvent molecule incorporation. It is also observed from Fig. 4 that the number of polar nitro groups (NO2) and amino groups (NH2) exposed on each surface is different, which will largely influence the interaction between the surface and acetic acid molecules.
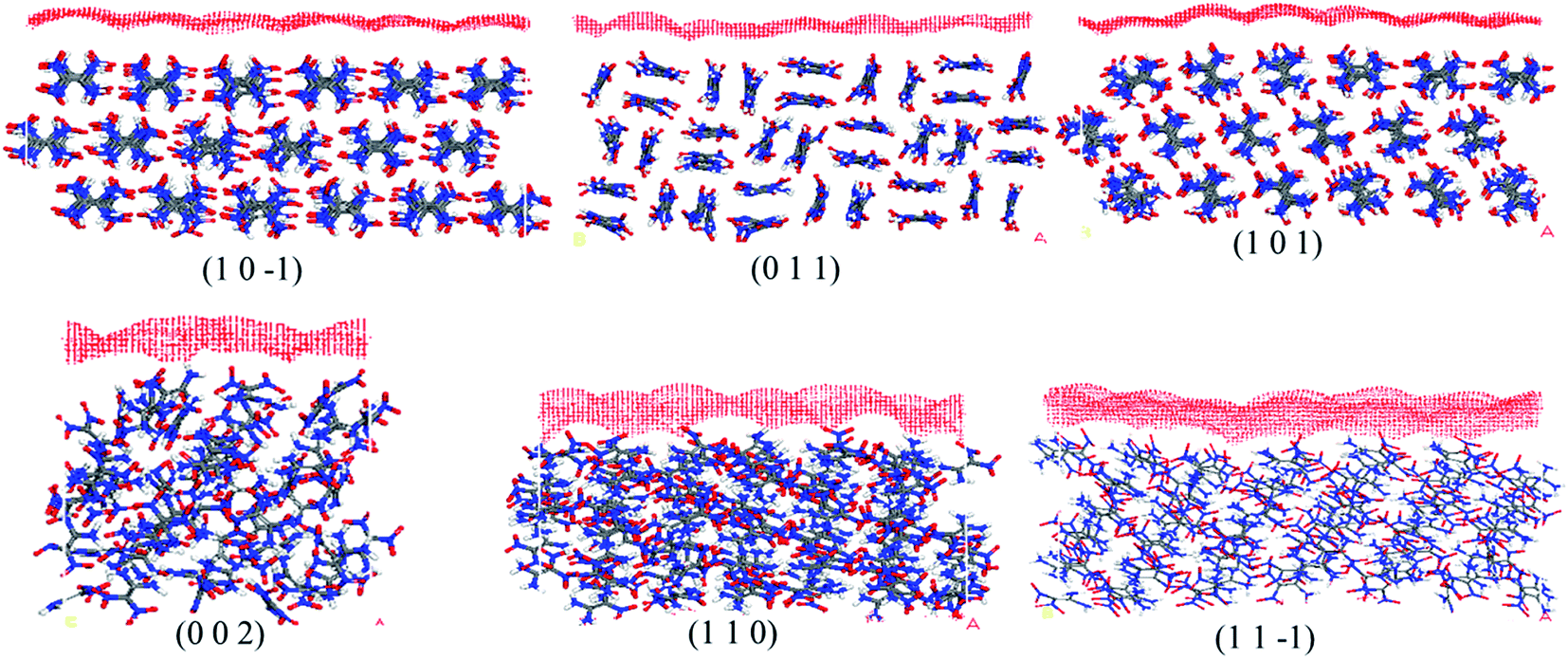 | ||
| Fig. 4 The geometric structures of different FOX-7 planes represented by the Connolly surface model. The red grid on the FOX-7 crystal face denotes the Connolly surface. | ||
| (h k l) | (1 0 −1) | (0 1 1) | (1 0 1) | (0 0 2) | (1 1 0) | (1 1 −1) |
|---|---|---|---|---|---|---|
| a A hkl is the area of the (h k l) face. Area unit is Å2. | ||||||
| A acc | 96.482 | 101.096 | 99.218 | 55.519 | 132.440 | 147.976 |
| A hkl | 87.827 | 91.184 | 88.665 | 46.061 | 108.898 | 117.925 |
| S | 1.10 | 1.11 | 1.12 | 1.21 | 1.22 | 1.25 |
Solvent–surface adsorption interactions are essential intermolecular interactions and can be divided into three types, including short-range interactions (hydrogen bonding and vdW interactions) and long-range interactions (Coulomb interactions). In order to analyze the solvent–crystal interactions clearly, the radial distribution functions (RDF) between acetic acid and the (1 1 −1) and (0 0 2) faces of FOX-7 were plotted as shown in Fig. 5. In the RDF graphs g(r) − r, the effective distances for hydrogen bonding, vdW interactions and Coulomb interactions are 2.6–3.5 Å, 3.5–5.5 Å and above 5.5 Å, respectively. Only the outermost layer of FOX-7 molecules which was nearest to the solvent was considered in the following analysis. From Fig. 5a, it can be seen that there high peaks exist in the range of 2.9–5.5 Å and beyond the 5.5 Å region, indicating the existence of hydrogen bonds and weak vdW interactions as well as electrostatic interactions. Similar phenomena can be observed when analyzing other pairs of atoms as shown in Fig. 5b and c, as well as in Fig. 5d–f. Moreover, the contributions from the interactions of Hsurface–Oacetic acid, Nsurface–Hacetic acid and Osurface–Hacetic acid in different systems are different. Meanwhile, Fig. 5c and f display the RDFs of Osurface–Hacetic acid in the two systems. From Fig. 5c, one can see that the higher peaks appear beyond 6.10 Å, which indicates that the systems mainly contain long-range interactions. Meanwhile for the interfacial model of the (0 1 1) surface and acetic acid, as shown in Fig. 5f, higher peaks also appear in the range of 3.10–5.5 Å except beyond the 7.70 Å region, representing strong short-range interactions. A similar conclusion that the contributions from the same pair of atoms in different systems are different can be found in other systems comprising acetic acid and the other crystal faces. That is to say, the solvent effect is determined by both the adsorption interactions between crystal surface and acetic acid and the surface structures. The modified attachment energies of the FOX-7 habit faces can be obtained using eqn (4), and the results are shown in Table 4.
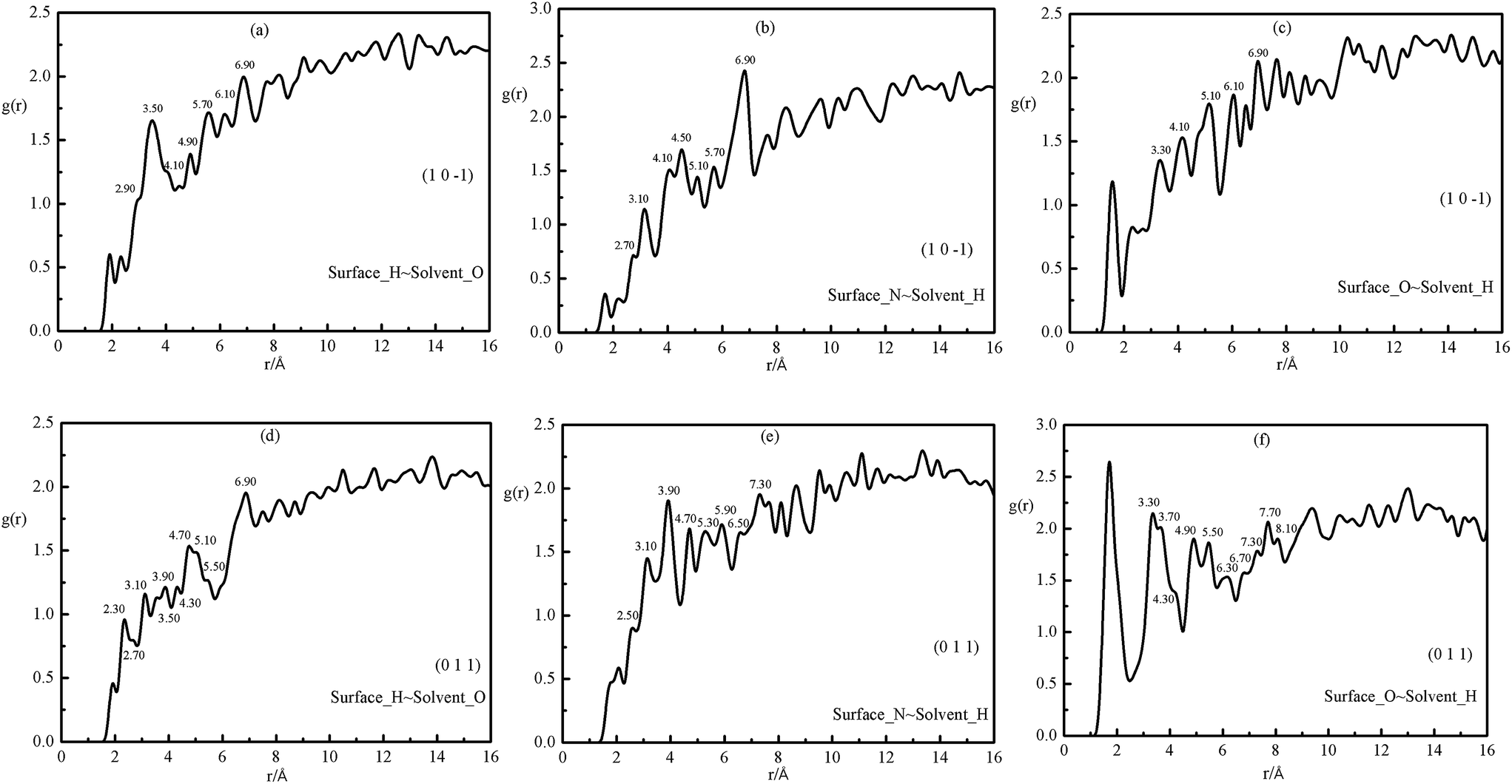 | ||
| Fig. 5 RDFs between acetic acid and FOX-7 atoms for the (1 0 −1) and (0 1 1) face adsorption systems. | ||
| (h k l) | E att hkl | R vac hkl | E s hkl | A acc | A box | E att,m hkl | R sol hkl |
|---|---|---|---|---|---|---|---|
| a All energies are in kcal mol−1, area unit is Å2. | |||||||
| (1 0 −1) | −28.03 | 1.00 | −135.85 | 96.482 | 790.441 | −11.44 | 1.00 |
| (0 1 1) | −30.63 | 1.09 | −206.62 | 101.096 | 820.653 | −5.17 | 0.45 |
| (1 1 −1) | −34.50 | 1.23 | −211.63 | 147.976 | 1061.330 | −5.00 | 0.44 |
| (1 0 1) | −37.15 | 1.33 | −246.88 | 99.219 | 797.983 | −6.45 | 0.56 |
| (1 1 0) | −39.91 | 1.42 | 50.57 | 132.44 | 980.081 | −46.74 | 4.08 |
| (0 0 2) | −43.60 | 1.56 | −150.52 | 55.519 | 414.55 | −23.44 | 2.05 |
The modified attachment energy quantitatively reflects the solvent effect. The larger attachment energy corresponds to the stronger effect of acetic acid on FOX-7 faces. Table 4 shows the modified attachment energies of FOX-7 habit faces. It can be found that the (1 1 −1) face has the minimum Eatt,mhkl of 5.00 kcal mol−1, while the (1 1 0) face has the maximum Eatt,mhkl of 46.74 kcal mol−1. The order of the modified attachment energies of the FOX-7 habit faces is (1 1 −1) < (0 1 1) < (1 0 1) < (1 0 −1) < (0 0 2) < (1 1 0), which is different from that in vacuum. Accordingly, the relative growth rates of the faces in vacuum and in acetic acid solvent have been calculated. Compared with those in vacuum, the growth rates of the (1 1 0) and (0 0 2) faces increase, while those of the (0 1 1), (1 1 −1) and (1 0 1) faces decrease. This indicates that the (1 1 −1) face should be of greatest morphological importance and the morphological importance of the (0 1 1) and (1 0 1) faces reduces, while that of the (1 0 −1), (0 0 2) and (1 1 0) faces disappears. Fig. 6a shows the FOX-7 crystal morphology grown from acetic acid solvent predicted by the MEA model, and the corresponding experimental shape is shown in Fig. 6b. It can be seen that the MEA model prediction is in good agreement with the shape observed experimentally.
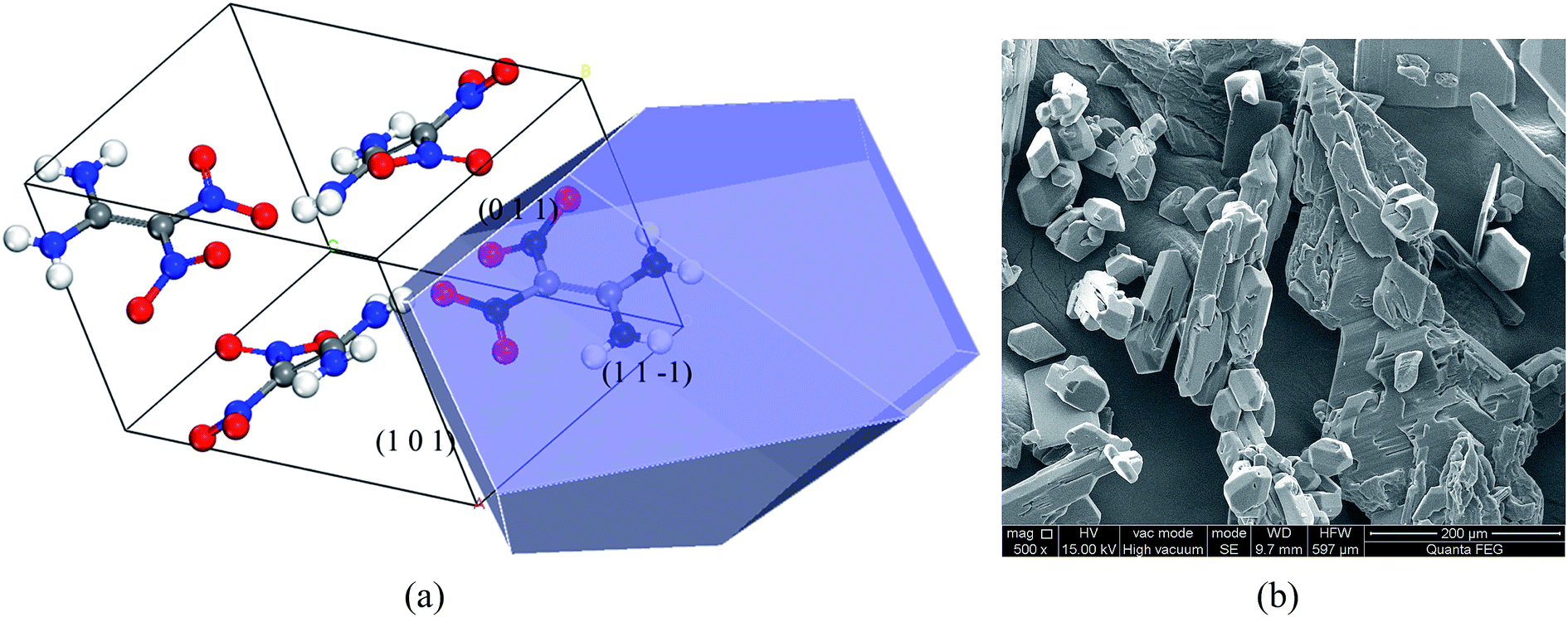 | ||
| Fig. 6 (a) FOX-7 crystal morphology grown from acetic acid solvent predicted by the MEA model, and (b) corresponding experimental shape. | ||
From the discussion on the crystal morphology of FOX-7 in acetic acid, one can see that the solvent effect depends on not only on adsorption interactions, but also on the surface structure. Using the MEA, predictions of the crystal morphology of FOX-7 in other solvents was performed. The adsorption energies for solvent adsorption on different FOX-7 surfaces, the modified attachment energies and the relative growth rates of FOX-7 habit faces are summarized in Table 5.
| Solvent | (h k l) | E att hkl | R vac hkl | E tot,avg hkl | E sur,avg hkl | E sol,avg hkl | E s hkl | A acc | S | E att,m hkl | R sol hkl |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a All energies are in kcal mol−1, area unit is Å2. b Standard deviations used as the estimated errors. | |||||||||||
| Cyclohexanone | (1 0 −1) | −28.03 | 1.00 | −26108.55 | −27524.44 | 1611.85 | −195.96(±13.22)b | 94.775 | 1.08 | −4.53 | 1.00 |
| (0 1 1) | −30.63 | 1.09 | −25948.94 | −27327.28 | 1606.38 | −228.04(±14.15) | 99.534 | 1.09 | −2.97 | 0.66 | |
| (1 1 −1) | −34.50 | 1.23 | −25826.46 | −27211.96 | 1654.33 | −268.83(±14.92) | 142.291 | 1.21 | 1.54 | — | |
| (1 0 1) | −37.15 | 1.33 | −26034.51 | −27413.91 | 1609.60 | −230.19(±12.44) | 97.404 | 1.10 | −9.05 | 2.00 | |
| (1 1 0) | −39.91 | 1.42 | −25889.28 | −27224.53 | 1639.88 | −304.63(±14.62) | 128.169 | 1.18 | −0.07 | 0.02 | |
| (0 0 2) | −43.60 | 1.56 | −12141.43 | −13566.50 | 1504.77 | −79.70(±14.97) | 54.072 | 1.17 | −33.20 | 7.33 | |
| Acetonitrile | (1 0 −1) | −28.03 | 1.00 | −28036.17 | −27524.44 | −363.47 | −148.26(±11.47) | 96.950 | 1.10 | −9.84 | 1.00 |
| (0 1 1) | −30.63 | 1.09 | −27872.37 | −27327.28 | −378.36 | −166.74(±12.49) | 101.504 | 1.11 | −10.00 | 1.02 | |
| (1 1 −1) | −34.50 | 1.23 | −27778.33 | −27211.96 | −361.33 | −205.04(±13.84) | 149.488 | 1.27 | −5.62 | 0.57 | |
| (1 0 1) | −37.15 | 1.33 | −27981.09 | −27413.91 | −382.70 | −184.48(±11.79) | 99.686 | 1.12 | −14.10 | 1.43 | |
| (1 1 0) | −39.91 | 1.42 | −27826.13 | −27224.53 | −342.05 | −259.55(±13.06) | 133.575 | 1.23 | −4.53 | 0.46 | |
| (0 0 2) | −43.60 | 1.56 | −14210.32 | −13566.50 | −446.88 | −196.95(±9.49) | 59.033 | 1.28 | −15.55 | 1.58 | |
| H2O/DMF | (1 0 −1) | −28.03 | 1.00 | −26127.46 | −27524.44 | 1618.50 | −221.52(±15.18) | 94.988 | 1.08 | −1.41 | 1.00 |
| (0 1 1) | −30.63 | 1.09 | −25928.79 | −27327.28 | 1640.65 | −242.16(±13.49) | 99.727 | 1.09 | −1.20 | 0.85 | |
| (1 1 −1) | −34.50 | 1.23 | −25812.49 | −27211.96 | 1696.65 | −297.18(±14.80) | 143.034 | 1.21 | 5.55 | — | |
| (1 0 1) | −37.15 | 1.33 | −26029.78 | −27413.91 | 1630.04 | −245.91(±17.92) | 97.643 | 1.10 | −7.06 | 5.02 | |
| (1 1 0) | −39.91 | 1.42 | −25835.24 | −27224.53 | 1688.20 | −298.92(±14.02) | 128.749 | 1.18 | −0.64 | 0.46 | |
| (0 0 2) | −43.60 | 1.56 | −12267.01 | −13566.50 | 1536.56 | −237.07(±13.34) | 55.418 | 1.20 | −11.90 | 8.46 | |
| H2O/DMSO | (1 0 −1) | −28.03 | 1.00 | −28091.33 | −27524.44 | −419.53 | −147.36(±12.13) | 95.064 | 1.08 | −10.30 | 1.00 |
| (0 1 1) | −30.63 | 1.09 | −27877.16 | −27327.28 | −389.73 | −160.16(±15.61) | 99.793 | 1.09 | −11.15 | 1.08 | |
| (1 1 −1) | −34.50 | 1.23 | −27773.37 | −27211.96 | −342.65 | −218.75(±13.07) | 143.309 | 1.22 | −4.97 | 0.48 | |
| (1 0 1) | −37.15 | 1.33 | −28019.25 | −27413.91 | −424.49 | −180.85(±15.76) | 97.724 | 1.10 | −15.00 | 1.46 | |
| (1 1 0) | −39.91 | 1.42 | −27752.35 | −27224.53 | −375.57 | −152.25(±13.44) | 128.943 | 1.18 | −19.88 | 1.93 | |
| (0 0 2) | −43.60 | 1.56 | −14173.14 | −13566.50 | −467.72 | −138.92(±11.06) | 55.558 | 1.21 | −24.98 | 2.42 | |
For cyclohexane adsorption on FOX-7 crystal surfaces, as seen from Table 5, it is found that the (1 1 0) face has the largest adsorption energy of −304.63 kcal mol−1, while the (0 0 2) face has the least value of −79.70 kcal mol−1. The rank of adsorption energies of the different FOX-7 growth faces can be written as follows: (1 1 0) > (1 1 −1) > (1 0 1) > (0 1 1) > (1 0 −1) > (0 0 2). Also, from Table 5 one can see that among all the FOX-7 habit faces, the (1 1 −1) face has the maximum S value of 1.21, indicating a rougher topography of the (1 1 −1) face, which is more convenient for solute or solvent molecule incorporation. The order of the modified attachment energies of the FOX-7 habit faces is (0 0 2) > (1 0 1) > (1 0 −1) > (0 1 1) > (1 1 0) > (1 1 −1), and the corresponding relative growth rates of FOX-7 habit faces in cyclohexanone solvent are listed. Compared with those in vacuum, the relative rates of the (0 1 1) and (1 1 0) faces decrease and the relative growth rate of the (1 1 0) face becomes the slowest. Conversely, the relative rates of the (1 0 1) and (0 0 2) faces increase. This indicates that due to the effect of cyclohexanone, the (1 1 0) face is of the greatest morphological importance and the (0 1 1) face is an important face, while other faces are not. The crystal morphology of FOX-7 grown from cyclohexanone solvent predicted by the MEA model is shown in Fig. 7a, and the corresponding experimental shape obtained by cooling crystallization is shown in Fig. 7b. It can be seen that the MEA model predictions agree well with the shape observed experimentally.
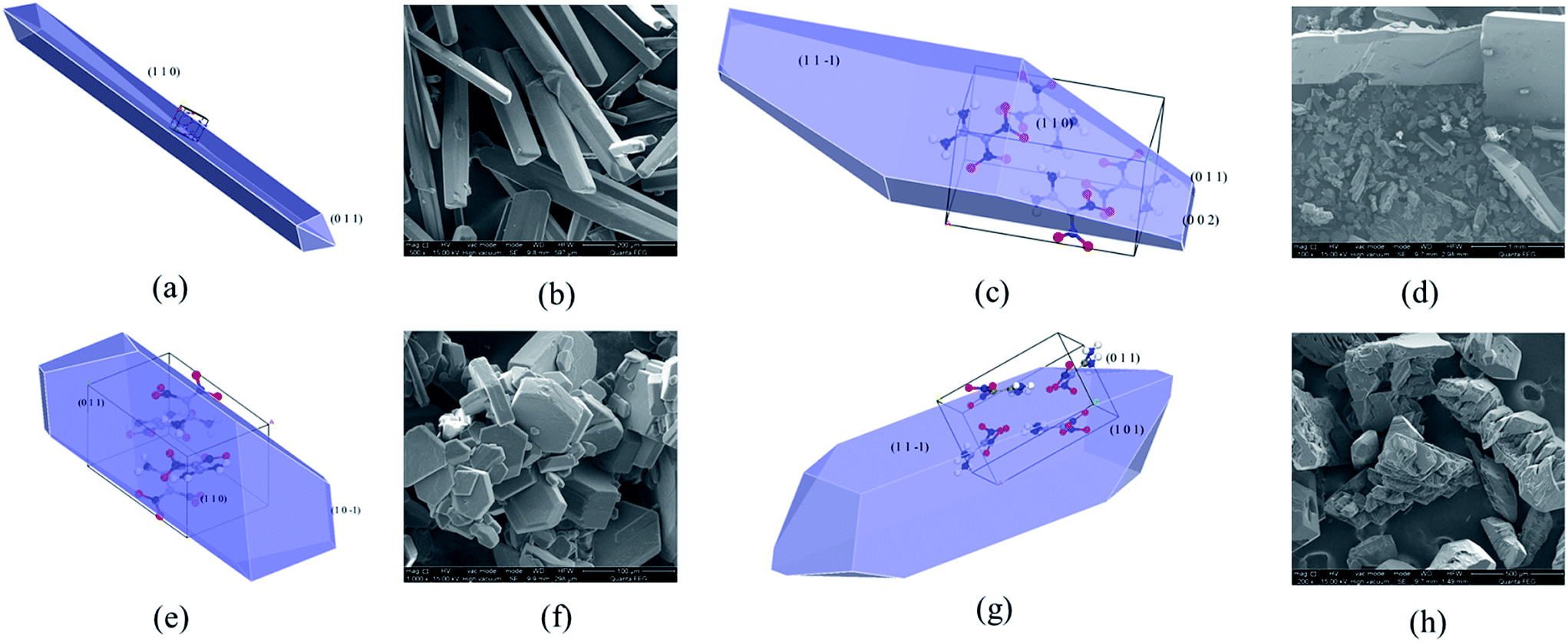 | ||
| Fig. 7 FOX-7 crystal morphology grown from cyclohexanone (a), acetonitrile (c), H2O/DMF (e), and H2O/DMSO (g) predicted by the MEA model; (b), (d), (f), and (h) are the corresponding experimental shape, respectively. | ||
For the system of FOX-7 crystal faces grown from acetonitrile solvent, it can be found that the interactions between the acetonitrile solvent and different crystal faces can be compared in the following sequence: (1 1 0) > (1 1 −1) > (0 0 2) > (1 0 1) > (0 1 1) > (1 0 −1). By considering the surface structures, the modified attachment energies of FOX-7 change in the order of (0 0 2) > (1 0 1) > (0 1 1) > (1 0 −1) > (1 1 −1) > (1 1 0), which suggests that in acetonitrile solvent the FOX-7 crystal would grow more rapidly in the (0 0 2) direction, while slowly in the (1 1 0) direction. Compared with that in vacuum, the relative growth rate of the (1 0 1) face largely increases, while that of the (0 0 2) face is almost unchanged. The final crystal morphology in acetonitrile calculated based on the modified attachment energy model is depicted in Fig. 7c, in which the (1 1 −1) and (1 1 0) faces are the most important faces. The prediction has been validated by experimental observations as shown in Fig. 7d.
Predictions of crystal morphologies of FOX-7 in mixed solvent such as H2O/DMF and H2O/DMSO were performed using the MEA model. For the system of FOX-7 growth from the H2O/DMF solution, the relative growth rates of the (1 0 1) and (0 0 2) faces are largely increased and those of the (0 1 1) and (1 1 0) faces are decreased. The morphological importance of the (0 1 1) and (1 1 0) faces become the greatest, while that of the (1 0 1) and (0 0 2) faces diminishes due to their weaker H2O/DMF solvent effect. The predicted morphology of FOX-7 in H2O/DMF is displayed in Fig. 7e, and the corresponding experimental shape is shown in Fig. 7f. The predicted FOX-7 morphology is in agreement with the experimental results.
If FOX-7 crystallizes from H2O/DMSO solvent, then solvent effects have brought about a significant change in the growth rates of the surfaces. It can be found that the descending order of interaction between H2O/DMSO solvent and different crystal faces is (1 1 −1) > (1 0 1) > (0 1 1) > (1 1 0) > (1 0 −1)> (0 0 2). By considering the surface structures, the rank of modified attachment energies of FOX-7 habit faces in H2O/DMSO solvent becomes (0 0 2) > (1 1 0) > (1 0 1) > (0 1 1) > (1 0 −1) > (1 1 −1), which is different from that in vacuum. This indicates that H2O/DMSO solvent has an important effect on FOX-7 crystal growth. Compared with that in vacuum, the relative growth rate of the (0 0 2) and (1 1 0) faces largely increases, while that of the (1 1 −1) face largely decreases. The final crystal morphology in H2O/DMSO calculated based on the modified attachment energy model is depicted in Fig. 7g, in which the (1 1 −1) face is the most important face. The predicted morphology is in reasonable agreement with the experimental results, as shown as Fig. 7h.
Compared with that in vacuum, the crystal morphologies of FOX-7 grown from solvents are different, which indicates that solvent largely influences the crystal morphology. The crystal morphologies of FOX-7 are variable in different crystallization solvents. The crystal shape of FOX-7 grown from cyclohexanone is similar to a prism, which shows the highest impact sensitivity. In contrast, the crystal shapes of FOX-7 grown from H2O/DMF and H2O/DMSO are regular, which are good choices for the solvent recrystallization of FOX-7.
5. Conclusions
We predicted the crystal morphologies of FOX-7 in different solvent using the modified attachment energy model incorporating the solvent effect. The calculated results show that the crystal of FOX-7 in vacuum predicted by AE model is dominated by six faces, (0 1 1), (1 0 −1), (1 0 1), (1 1 −1), (0 0 2) and (1 10), which is related to the strength of the bond energies between FOX-7 molecules. Taking the system of acetic acid adsorbed on FOX-7 surfaces as an example, the solvent effect on the crystal morphology of FOX-7 was investigated. The solvent molecules adsorb on the FOX-7 faces mainly via hydrogen bonding and vdW interactions, as well as Coulomb interactions. The forces vary on different surfaces. When taking the term of solvent effect into account, the modified attachment energy changes. The (1 1 −1), (0 1 1) and (1 0 1) faces have morphological importance.The model has been successfully extended to the study of crystal FOX-7 grown from cyclohexanone, acetonitrile, H2O/DMF, and H2O/DMSO. The model is validated by the experimental results. The crystal morphologies of FOX-7 are variable in different crystallization solvents. H2O/DMF and H2O/DMSO are good choices for the solvent recrystallization of FOX-7. The results can provide some theoretical support for explosive morphology control technology.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No: 21473108) and the Fundamental Research Funds for Shaanxi Innovative Team of Key Science and Technology (2013KCT-17).Notes and references
- M. Anniyappan, M. B. Talawar, G. M. Gore, S. Venugopalan and B. R. Gandhe, J. Hazard. Mater., 2006, 137, 812–819 CrossRef CAS PubMed.
- N. V. Latypov, M. Johansson, E. Holmgren, E. V. Sizova, V. V. Sizov and A. J. Bellamy, Org. Process Res. Dev., 2007, 11, 56–59 CrossRef CAS.
- J. Evers, T. M. Klapötke, P. Mayer, G. Oehlinger and J. Welch, Inorg. Chem., 2006, 45, 4996–5007 CrossRef CAS PubMed.
- M.-J. Crawford, J. Evers, M. Göbel, T. M. Klapötke, P. Mayer, G. Oehlinger and J. M. Welch, Propellants, Explos., Pyrotech., 2007, 32, 478–495 CrossRef CAS.
- M. M. Bishop, N. Velisavljevic, R. Chellappa and Y. K. Vohra, J. Phys. Chem. A, 2015, 119, 9739–9747 CrossRef CAS PubMed.
- M. M. Bishop, R. S. Chellappa, M. Pravica, J. Coe, Z. Liu, D. Dattlebaum, Y. Vohra and N. Velisavljevic, J. Chem. Phys., 2012, 137, 174304 CrossRef PubMed.
- Z. A. Dreger, Y. Tao and Y. M. Gupta, Chem. Phys. Lett., 2013, 584, 83–87 CrossRef CAS.
- H.-X. Gao, F.-Q. Zhao, R.-Z. Hu, Q. Pan, B.-Z. Wang, X.-W. Yang, Y. Gao and S.-L. Gao, Chin. J. Chem., 2006, 24, 177–181 CrossRef CAS.
- K. Xu, J. Song, F. Zhao, H. Ma, H. Gao, C. Chang, Y. Ren and R. Hu, J. Hazard. Mater., 2008, 158, 333–339 CrossRef CAS PubMed.
- X.-L. Xing, L. Xue, F.-Q. Zhao, H.-X. Gao and R.-Z. Hu, Thermochim. Acta, 2009, 491, 35–38 CrossRef CAS.
- B. Yuan, Z. Yu and E. R. Bernstein, J. Chem. Phys., 2014, 140, 074708 CrossRef PubMed.
- W. A. Trzciński, S. Cudziło, Z. Chyłek and L. Szymańczyk, J. Hazard. Mater., 2008, 157, 605–612 CrossRef PubMed.
- A. K. Mandal, U. Thanigaivelan, R. K. Pandey, S. Asthana, R. B. Khomane and B. D. Kulkarni, Org. Process Res. Dev., 2012, 16, 1711–1716 CrossRef CAS.
- B. Gao, P. Wu, B. Huang, J. Wang, Z. Qiao, G. Yang and F. Nie, New J. Chem., 2014, 38, 2334–2341 RSC.
- Z. A. Dreger, A. I. Stash, Z.-G. Yu, Y.-S. Chen, Y. Tao and Y. M. Gupta, J. Phys. Chem. C, 2016, 120, 1218–1224 CrossRef CAS.
- D. E. Taylor, F. Rob, B. M. Rice, R. Podeszwa and K. Szalewicz, Phys. Chem. Chem. Phys., 2011, 13, 16629–16636 RSC.
- U. Teipel, Energetic Materials: Particle Processing and Characterization, John Wiley & Sons, 2006 Search PubMed.
- T. M. Tillotson, L. W. Hrubesh, R. L. Simpson, R. S. Lee, R. W. Swansiger and L. R. Simpson, J. Non-Cryst. Solids, 1998, 225, 358–363 CrossRef CAS.
- J.-W. Kim, D. B. Park, H.-M. Shim, H.-S. Kim and K.-K. Koo, Ind. Eng. Chem. Res., 2012, 51, 3758–3765 CrossRef CAS.
- J. Chen, J. Wang, J. Ulrich, Q. Yin and L. Xue, Cryst. Growth Des., 2008, 8, 1490–1494 Search PubMed.
- M. N. Bhat and S. M. Dharmaprakash, J. Cryst. Growth, 2002, 242, 245–252 CrossRef CAS.
- M. Lahav and L. Leiserowitz, Chem. Eng. Sci., 2001, 56, 2245–2253 CrossRef CAS.
- C. Thompson, M. C. Davies, C. J. Roberts, S. J. B. Tendler and M. J. Wilkinson, Int. J. Pharm., 2004, 280, 137–150 CrossRef CAS PubMed.
- M. J. Siegfried and K.-S. Choi, J. Am. Chem. Soc., 2006, 128, 10356–10357 CrossRef CAS PubMed.
- X. Mao, X. Song, G. Lu, Y. Xu, Y. Sun and J. Yu, Chem. Eng. J., 2015, 278, 320–327 CrossRef CAS.
- R. J. Hudson, M. Moniruzzaman and P. P. Gill, Propellants, Explos., Pyrotech., 2015, 40, 233–237 CrossRef CAS.
- Z. Zhang, Y. Liu, Y. Yang and B. I. Yakobson, Nano Lett., 2016, 16, 1398–1403 CrossRef CAS PubMed.
- E. Kanaki, S. Gohr, C. Müller and B. Paulus, Surf. Sci., 2015, 632, 158–163 CrossRef CAS.
- X. Duan, C. Wei, Y. Liu and C. Pei, J. Hazard. Mater., 2010, 174, 175–180 CrossRef CAS PubMed.
- W. Shi, M. Xia, W. Lei and F. Wang, J. Mol. Graphics Modell., 2014, 50, 71–77 CrossRef CAS PubMed.
- G. Chen, M. Xia, W. Lei, F. Wang and X. Gong, J. Phys. Chem. A, 2014, 118, 11471–11478 CrossRef CAS PubMed.
- G. Chen, C. Chen, M. Xia, W. Lei, F. Wang and X. Gong, RSC Adv., 2015, 5, 25581–25589 RSC.
- H.-E. Lee, T. B. Lee, H.-S. Kim and K.-K. Koo, Cryst. Growth Des., 2010, 10, 618–625 Search PubMed.
- C. Zhang, C. Ji, H. Li, Y. Zhou, J. Xu, R. Xu, J. Li and Y. Luo, Cryst. Growth Des., 2013, 13, 282–290 Search PubMed.
- H.-M. Shim, H.-S. Kim and K.-K. Koo, Cryst. Growth Des., 2015, 15, 1833–1842 Search PubMed.
- W. Qian, C.-Y. Zhang, H.-H. Zong, Y. Xiong, W.-B. Zhang and Y.-J. Shu, J. At. Mol. Phys., 2014, 31, 454–462 CAS.
- X.-T. Ren, D.-Y. Ye, N. Ding, J.-X. He, Y.-H. Lu, Q. Lei and Y.-Y. Guo, Acta Armamentarii, 2015, 36, 272–278 Search PubMed.
- P. Hartman and P. Bennema, J. Cryst. Growth, 1980, 49, 145–156 CrossRef CAS.
- Z. Berkovitch-Yellin, J. Am. Chem. Soc., 1985, 107, 8239–8253 CrossRef CAS.
- H. J. Leamy, G. H. Gilmer, K. A. Jackson and J. M. Blakely, Surface Physics of Materials, Academic Press, 1975 Search PubMed.
- Material Studio 5.5, Acceryls Inc., San Diego, 2010 Search PubMed.
- R. Gilardi, CCDC 127539: Experimental Crystal Structure Determination, Cambridge Crystallographic Data Centre, Cambridge, U.K., 1999.https://summary.ccdc.cam.ac.uk/structure-summary?ccdc=127539.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- H. Fan, K. Zhang and M. M. Yuen, Thermal Performance of Carbon Nanotube-Based Composites Investigated by MolecularDynamics Simulation, Electronic Components and Technology Conference, 2007, ECTC’07, Proceedings. 57th, IEEE, 2007, pp. 269–272 Search PubMed.
- D. C. Sorescu, J. A. Boatz and D. L. Thompson, J. Phys. Chem. A, 2001, 105, 5010–5021 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- M. Connolly, Science, 1983, 221, 709–713 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 157–167 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed . Multiwfn official website: http://Multiwfn.codeplex.com.
- Q. Zhou, Z. Chen, C. Zheng, B. Wang, X. Ren, K. Wang, M. Wang and C. Zhou, Chin. J. Explos. Propellants, 2014, 37, 67–69 CAS.
| This journal is © The Royal Society of Chemistry 2016 |