DOI:
10.1039/C6RA06833B
(Paper)
RSC Adv., 2016,
6, 44261-44271
Carbon rings: a DFT study on geometry, aromaticity, intermolecular carbon–carbon interactions and stability†
Received
15th March 2016
, Accepted 27th April 2016
First published on 28th April 2016
Abstract
Non-covalent dimer formation and intermolecular bonding features of planar monocyclic carbon rings showing C4N+2 and C4N configurations have been studied using the meta-GGA DFT method, M06L/6-311+G(d) for N = 1–8. The C4N+2 show cumulenic structures with equal bond lengths and C4N form structures with clear bond length alternation. The doubly Hückel aromatic nature of C4N+2 is revealed through two cyclic delocalized π-molecular orbitals and highly negative nucleus independent chemical shift (NICS) parameters while the doubly Hückel antiaromatic nature of C4N is brought out through two localized π-molecular orbitals and highly positive NICS parameters. Further, the uniform electron distribution over the delocalized CC bonds in C4N+2 and the alternate electron rich and electron deficient regions in C4N are assessed on the basis of the critical features of the molecular electrostatic potential (MESP). The contrasting geometric, electronic and magnetic features of C4N+2 compared to C4N result in a drastic difference in their intermolecular bonding behaviour. The C4N showed a much higher tendency than C4N+2 for dimer formation as the former, in general show a 4N number of intermolecular C⋯C interactions due to complimentary electrostatic interactions between electron rich shorter CC bonds and electron deficient longer CC bonds. In C4N dimers, a perfect sandwich configuration is preferred to maximize the attractive complementary electrostatic interactions while in C4N+2 dimers a shifted-parallel stacked arrangement indicated the non-complementary character of interactions arising from smooth aromatic distribution of electrons. The comparative stability of the carbon rings and unsubstituted polyynes is quantified by measuring the homodesmotic reaction energy (Ehdr) with acetylene. The Ehdr indicated significant stabilization of C4N+2 compared to C4N. The energy required to open up a carbon ring to the linear form is computed as Eopening and this quantity is used to estimate the aromatic stabilization of C4N+2 as well as the antiaromatic destabilization of C4N systems.
Introduction
Laser vaporization of graphite results in the formation of carbon clusters.1 Depending on the size of the cluster, several types of carbon clusters exist such as linear chains, rings, bowls, plates and cages.2–6 Very small clusters such as C2 and C3 exist as linear chains. At larger sizes, they form rings. Carbon rings are stable in the size range of approximately C6–C30, which show slight variation with the theoretical methods used as well as the experimental conditions. At even higher sizes, these high energy sp hybridized clusters start converting into sp2 hybridized forms such as plates and cages. These structures rearrange to form fullerenes, the mechanism of which has been studied extensively.7–13 The ring structures are relatively stable non spheroidal forms. Among the ring structures, the monocyclic rings are relatively more stable compared to other configurations.14
The properties of planar monocyclic carbon rings have been extensively studied theoretically as well as experimentally.2,5,6,14–27 The properties of these rings vary with the number of carbon atoms, n depending on whether n is odd, n = 4N or n = 4N + 2 where N is an integer. The stabilization of this type of molecules in different structural types has been explained in terms of aromaticity, second order Jahn–Teller distortions and Peierls instability effects.27 The structures reported by Torelli and Mitas for the molecules with 4N + 2 carbon atoms using quantum Monte Carlo methods include (i) rings with all bond lengths and bond angles equal (Dnh symmetry), (ii) rings with alternating bond angles (Dn2h symmetry) or (iii) rings with alternating bond lengths (Dn2h symmetry).27 The concept of double aromaticity28 was first coined by Schleyer et al. in 1979.29 In the rings with 4N + 2 carbon atoms, the presence of two sets of conjugated π electron systems (in plane and out of plane) suggests double aromaticity. Theoretical studies have shown that, at smaller sizes, carbon rings with 4N + 2 carbon atoms show double aromaticity whereas those with 4N carbon atoms show double antiaromaticity.30–32 The diatropic and paratropic ring currents in both the delocalized π electron systems of C4N+2 and C4N molecules respectively have been described well by Fowler et al.33 using the maps of current density. At very large sizes, both types of rings are expected to show non aromatic behaviour.30
The structure for a carbon ring Cn is predicted to be cumulenic with bond angle alternation if n = 4N + 2 (type (ii)) and acetylenic with bond length alternation if n = 4N (type (iii)) while both maintains a Dn2h symmetry.25–27,30,33,34
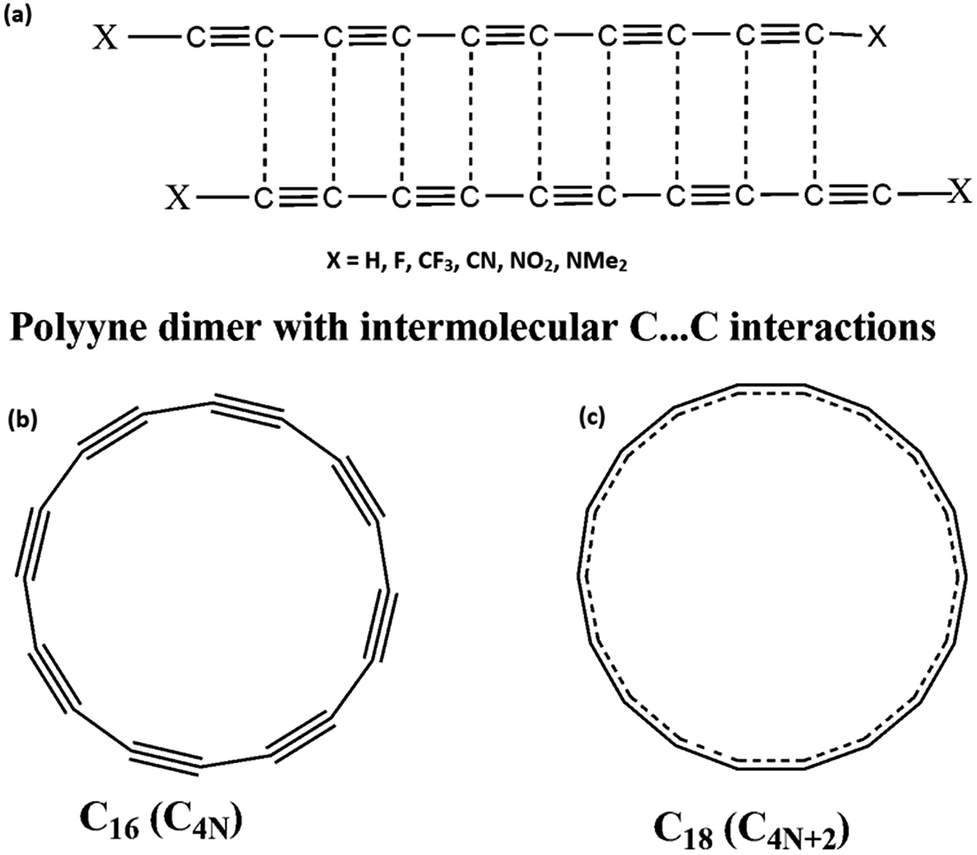
Here, we focus mainly on the intermolecular interactions in planar monocyclic carbon rings. In a previous study, we have shown that inter-molecular C⋯C interactions between carbon atoms in similar chemical environments exist in several dipolar organic molecules.35 These interactions result from complimentary electrostatic interaction of electron rich region of one molecule with electron deficient region of another. For instance, in the case of polyyne molecules, the electron rich formal triple bond of one molecule interacts with electron deficient formal single bond of another36 (Scheme 1) and several such interactions along the polyyne chain lead to significant stabilization of the dimer, approximately 1.00 kcal mol−1 for each C⋯C interactions. Natural bond orbital (NBO) analysis showed both the donor and acceptor character of the interacting carbon atoms (i.e., in a C1⋯C2 interaction, a charge transfer from C1 to C2 is also complimented by a similar charge transfer from C2 to C1, where C1 and C2 are chemically almost identical).35
 |
| | Scheme 1 (a) Polyyne dimers showing intermolecular C⋯C interactions (b) C16 with acetylenic structure and (c) C18 with cumulenic structure. | |
In polyynes, though the interacting carbon atoms belong to similar chemical environments, local differences in electron concentration result in the C⋯C interactions. The end substitutions, which affected the electron distribution throughout the length of the polyyne molecules, had a strong influence on the strength of the C⋯C interactions.36 In the case of Cn rings, intermolecular interactions could not be varied by substitutions as such manipulations are not possible for them. The only option is to vary the electronic configuration of the ring structure. As illustrated in Scheme 1, the C4N rings have similarity with polyyne molecules as they possess alternate electron rich and electron deficient regions which would lead to several intermolecular C⋯C interactions. On the other hand, in C4N+2 molecules, all the CC bonds are identical due to aromatic π-electron delocalization and the chance of seeing several C⋯C interactions is less. For testing this hypothesis, dimers of Cn molecules are studied for n = 4N + 2 and n = 4N, where N is a natural number. First, we illustrate the doubly aromatic stabilization of C4N+2 molecules and anti-aromatic nature of C4N molecules using their geometric, molecular electrostatic potential (MESP) and magnetic features for C6 to C32 molecules and then study the intermolecular C⋯C interactions in them. A study of aromatic and antiaromatic character of planar carbon rings based on their MESP features and their intermolecular bonding behaviour have not been reported in the literature. Further, the stability of these Cn molecules is compared with polyynes using homodesmotic reactions to predict their existence. Finally, ring opening reaction leading to chain structures is examined to assess the aromatic stabilization or antiaromatic destabilization of the systems.
Computational methods
The Cn molecules with n = 4N + 2 and n = 4N are optimized for n varying from 6 to 32. Dimers of molecules up to C28 are also optimized. All the molecules are optimized using the meta-GGA density functional M06L37 with the basis set 6-311++g(d,p). This method is shown to be very good in the study of non-covalently interacting system in an extensive benchmark study38 followed by several other studies35,36,39,40 where the results given by this method are confirmed using many reliable DFT and ab initio methods. The geometries are confirmed to be minima by frequency calculation. All the calculations have been done using Gaussian09![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41 suit of programs. Interaction energy (Eint) of a dimer is calculated by subtracting twice the energy of an isolated monomer from the energy of the dimer. Basis set superposition energy (BSSE) is calculated using Boys and Bernardi method42 as implemented in Gaussian09. Maps of molecular electrostatic potential (MESP)43 are used for illustrating the delocalized (in C4N+2) and localized (in C4N) nature of electrons. Also, the intermolecular interactions are explained using the MESP features of the dimers. MESP features have been used for understanding several phenomena in chemistry44,45 including different types of intermolecular interactions.35,36,46–48 The most negative value of MESP in a molecule is denoted as Vmin. The position and magnitude of Vmin can used for understanding the position and strength of electron rich regions such as lone pairs of molecules as well as their interactions with nucleophiles.49–52 Here, Vmin values are taken as indicators of localization/delocalization nature of π electrons in the carbon rings. Quantum theory of atoms-in-molecule (QTAIM) analysis as implemented in AIMAll software package53 is used for the study of intermolecular interactions. This analysis gives (3, −1) critical point, known as bond critical points (BCPs) between two bonded atoms. The value of electron density, ρ at a BCP is considered as a measure of the strength of that bonding interaction. Nucleus-independent chemical shift (NICS),54–56 which is the absolute magnetic shielding of the induced ring currents at the centre of rings is used for studying the aromatic/antiaromatic behaviour of a molecule. Negative value for NICS indicates aromaticity and positive value indicates antiaromaticity. Since the chemical shift values calculated at the centre of a ring namely NICS(0) can be contaminated by shielding contributions from the core as well as σ electrons, we have also calculated NICS(1),57 observed at 1 Å above the plane of the ring, where the effect of σ electrons is reduced and the π system is maximized.58
41 suit of programs. Interaction energy (Eint) of a dimer is calculated by subtracting twice the energy of an isolated monomer from the energy of the dimer. Basis set superposition energy (BSSE) is calculated using Boys and Bernardi method42 as implemented in Gaussian09. Maps of molecular electrostatic potential (MESP)43 are used for illustrating the delocalized (in C4N+2) and localized (in C4N) nature of electrons. Also, the intermolecular interactions are explained using the MESP features of the dimers. MESP features have been used for understanding several phenomena in chemistry44,45 including different types of intermolecular interactions.35,36,46–48 The most negative value of MESP in a molecule is denoted as Vmin. The position and magnitude of Vmin can used for understanding the position and strength of electron rich regions such as lone pairs of molecules as well as their interactions with nucleophiles.49–52 Here, Vmin values are taken as indicators of localization/delocalization nature of π electrons in the carbon rings. Quantum theory of atoms-in-molecule (QTAIM) analysis as implemented in AIMAll software package53 is used for the study of intermolecular interactions. This analysis gives (3, −1) critical point, known as bond critical points (BCPs) between two bonded atoms. The value of electron density, ρ at a BCP is considered as a measure of the strength of that bonding interaction. Nucleus-independent chemical shift (NICS),54–56 which is the absolute magnetic shielding of the induced ring currents at the centre of rings is used for studying the aromatic/antiaromatic behaviour of a molecule. Negative value for NICS indicates aromaticity and positive value indicates antiaromaticity. Since the chemical shift values calculated at the centre of a ring namely NICS(0) can be contaminated by shielding contributions from the core as well as σ electrons, we have also calculated NICS(1),57 observed at 1 Å above the plane of the ring, where the effect of σ electrons is reduced and the π system is maximized.58
Results and discussion
Geometry of the carbon rings
All C4N+2 molecules show a cumulenic structure with all the bond lengths equal while all the C4N molecules show a clear bond length alternation with alternate shorter (formal triple) and longer (formal single) bonds. The cumulenic structure of a C4N+2 is indicative of a delocalized π-electron distribution and aromatic stabilization. The variation of average of all the CC bond lengths in C4N+2 molecules with n varying from 6 to 30 is represented in Fig. 1. The largest CC bond length (1.33 Å) is observed in C6, which is decreased to 1.29 Å in C10 and is almost constant (around 1.28 Å) in all other molecules. The C4N molecules show localized triple bonds, which is a characteristic feature of antiaromatic molecules.54 In C8 to C32, the longer (single) bond lengths vary in the range 1.39 to 1.32 Å and the shorter (triple) bonds vary from 1.27 to 1.24 Å. The variation of the average of single and triple bond lengths of all the C4N molecules with the value of n is also shown in Fig. 1. Both single and triple bond lengths first decrease and then become almost a constant.
 |
| | Fig. 1 Average bond lengths of the planar carbon rings (Cn). C4N+2 molecules show nearly equal lengths for all the C–C bonds indicating π-electron delocalization. The C4N molecules show alternating long and short bonds. | |
The bond angles are found to depend only on the ring size and not on the aromatic or antiaromatic behaviour of the molecules. To some extent, all the Cn molecules show bond angle alternation. The difference between two adjacent bond angles decreases with increase in ring size and the bond angles reaches almost the same value in larger rings. For instance, the two bond angles observed in C6 are 154° and 86° while in C32, the bond angles show only a small variation between 168° and 170°. In every C4N+2 and C4N systems, two symmetrically unequal sets of carbon atoms can be identified referred to herein as Cα and Cβ. The Cα atoms are characterized by larger CCC angles centred on them compared to Cβ. As the system size increases, the symmetrical inequality to distinguish Cα and Cβ decreases. This can be attributed to the tendency for bond angle equalization in C4N+2 leading to the formation of chemically identical environment for individual atoms as well as CC bonds. Whereas in C4N systems, bond angle equalization suggests the tendency of all the carbon atoms in the system to remain in identical chemical environment but the bond length alternation suggests the tendency to keep two kinds of bonds in the system.
Molecular electrostatic potential (MESP)
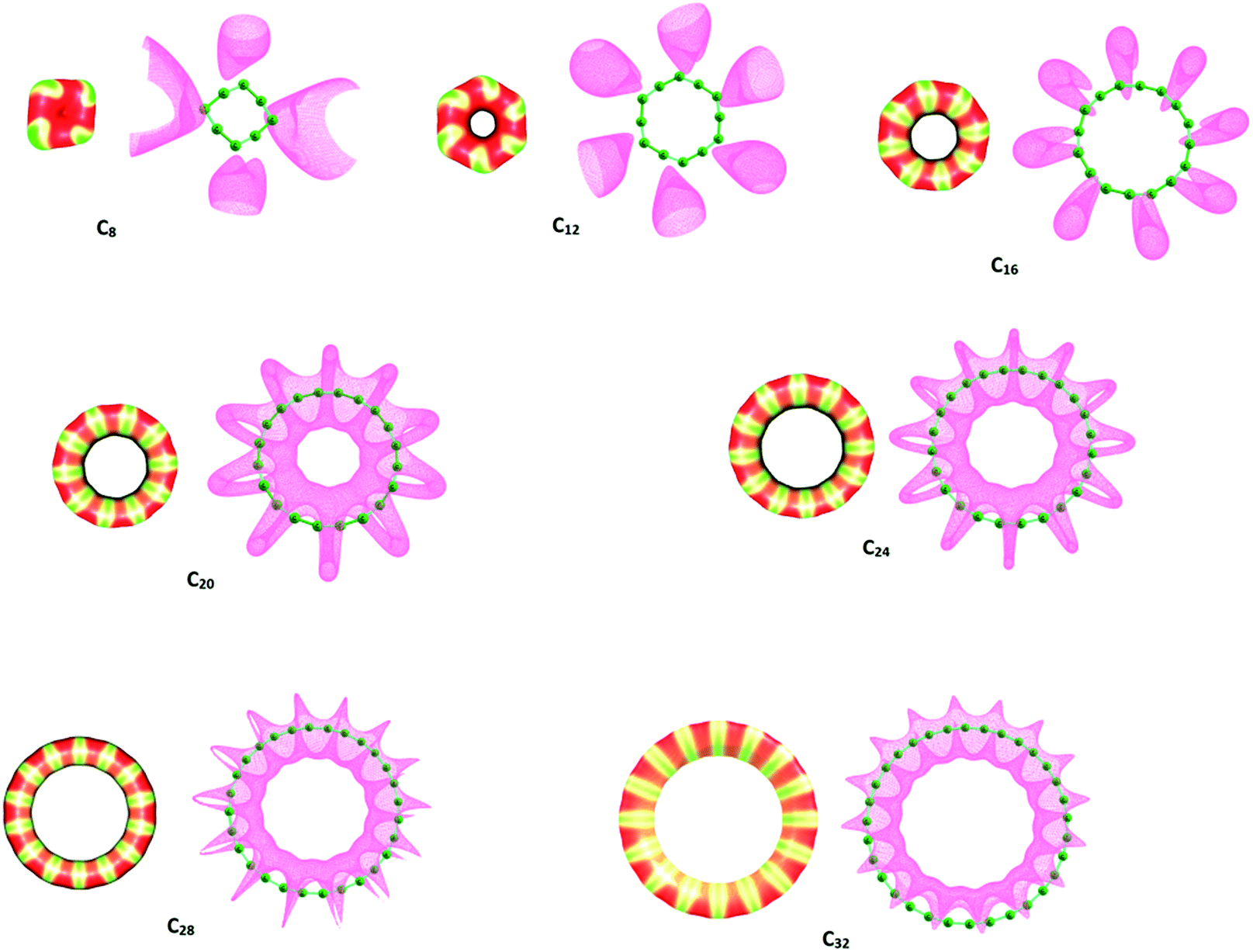
The MESP mapped on to 0.01 au electron density surface and MESP isosurface of −0.00075 au are shown in Fig. 2 for the C4N+2 molecules. The Vmin values of all the Cn systems are given in Table 1. The MESP map of C6 clearly shows that Cα atoms are electron deficient compared to the Cβ atoms. The MESP topography of C6 reveals a MESP minimum (Vmin) value −12.14 kcal mol−1 for the Cβ atoms, which has the highest magnitude for Vmin among all the Cn molecules studied. This indicates some amount of divalent carbene type character to these atoms due to underutilization of its electrons for π-conjugation. The inter-atomic distance between Cα carbon atoms is 1.809 Å which suggests weak interactions between them. The electron deficient nature of these carbon atoms could be attributed to the utilization of their electrons for such interactions. In C10, the electron rich Cβ shows Vmin value of −3.53 kcal mol−1, which is 8.63 kcal mol−1 less negative than C6, indicating increased delocalization of the π-electrons. In C14, the bond angle alternation at Cα and Cβ atoms is very small (about 10°) compared to C6 (about 68°) and C10 (about 38°) indicating a near perfect π-electron delocalization characteristic of aromatic systems. Unlike C6 and C10, the MESP isosurface map of C14 does not show significant electron localization in the molecular plane whereas negative-valued MESP appears above and below the ring plane. Such a feature identified as typical of a π-electron cloud is well-known in the case of benzene. Also, the lowest magnitude for Vmin among all the molecules is shown by C14, indicating highly delocalized nature of π electrons. Beyond C14, all higher members of C4N+2 series show the π-electron cloud. However, unlike C14, the larger rings show the influence of the π-electron in the interior central regions.
 |
| | Fig. 2 MESP mapped on to 0.01 au electron density isosurface (left) and MESP isosurface (pink-coloured) at −0.00075 au of C4N+2 molecules. Colour coding from blue to red indicates MESP values in the range −0.03 to 0.05 au. | |
Table 1 Values of the most negative electrostatic potential (Vmin) of C4N+2 and C4N rings in kcal mol−1
| C4N+2 |
Vmin |
C4N |
Vmin |
| C6 |
−12.14 |
C8 |
−11.81 |
| C10 |
−3.53 |
C12 |
−5.11 |
| C14 |
−0.54 |
C16 |
−2.35 |
| C18 |
−1.29 |
C20 |
−1.16 |
| C22 |
−1.39 |
C24 |
−1.24 |
| C26 |
−1.30 |
C28 |
−1.22 |
| C30 |
−1.17 |
C32 |
−1.27 |
In the case of C4N series, MESP features of C8 and C12 show electron rich character of Cβ compared to Cα (Fig. 3). The Vmin values of C8 and C12 are −11.81 and −5.11 kcal mol−1, respectively. In the case of C16 and other higher systems, the MESP isosurface is localized on the shorter CC bonds. The red-coloured regions in the MESP surface maps indicate electron deficient longer CC bonds. As the ring size increases, the colour contrasts indicating electron rich and electron deficient region decreases. Such an effect is more evident in C4N+2 systems than C4N indicating more delocalized distribution of electrons in the former than the latter. The MESP map also suggests that C4N+2 rings possess a cumulenic carbyne structure while C4N rings possess acetylenic carbyne structure. The acetylenic character of the latter can be correlated to the cylindrical distribution of π-electrons characterized by the MESP isosurface shaped like a ‘ring’ around the shorter CC bonds as seen in C20–C32.
 |
| | Fig. 3 MESP mapped on to 0.01 au electron density isosurface (left) and MESP isosurface (pink-coloured) at −0.00075 au of C4N molecules. Colour coding from blue to red indicates MESP values in the range −0.03 to 0.05 au. | |
Nucleus-independent chemical shift (NICS)
The NICS(0) and NICS(1) values of C4N+2 and C4N rings are listed in Table 2. The negative NICS values of C4N+2 clearly illustrate their aromatic behaviour. The high values of NICS in these rings support double aromaticity, viz. one due to the conjugation of the π-electron that lie in the σ-plane and the other due to the conjugation of π-electrons orthogonal to this plane. To illustrate this point, the two delocalized π-molecular orbitals of a representative C4N+2 system C18 is shown in Fig. 4. The C4N rings show positive NICS(0) and NICS(1), showing their anti-aromatic character. The high positive values of NICS may indicate the possibility of double antiaromatic character arising from 4N π-electrons in the σ-plane and 4N π-electrons in the plane perpendicular to the σ-plane. The localized nature of the two π-molecular orbitals of C16 given in Fig. 4 supports the double antiaromatic nature of C4N systems.
Table 2 NICS values in ppm of the C4N+2 and C4N rings
| C4N+2 |
NICS(0) |
NICS(1) |
C4N |
NICS(0) |
NICS(1) |
| C6 |
−22.64 |
−9.30 |
C8 |
44.21 |
36.39 |
| C10 |
−30.89 |
−22.82 |
C12 |
54.69 |
45.42 |
| C14 |
−40.53 |
−34.10 |
C16 |
50.31 |
44.10 |
| C18 |
−42.30 |
−37.94 |
C20 |
43.47 |
39.83 |
| C22 |
−42.54 |
−39.52 |
C24 |
37.49 |
35.25 |
| C26 |
−42.73 |
−40.51 |
C28 |
32.14 |
30.71 |
| C30 |
−42.87 |
−41.18 |
C32 |
28.26 |
27.29 |
 |
| | Fig. 4 The delocalized π-molecular orbitals of a C4N+2 system C18 (top) and the localized π-molecular orbitals of a C4N system C16 (bottom). | |
The magnitude of NICS(0) in C4N+2 increases with the ring size from C6–C18 and then remains almost constant up to C30. NICS(1) also follows almost similar trends. Although this data may suggest enhancement in aromaticity of a C4N+2 ring with increase in its size up to C18 and not much variation afterwards, a confirmation of this feature is difficult as NICS is ring size dependent. Therefore, we compare the NICS(0) value of C6 (−22.64) with that of benzene (−9.7 (ref. 54)), which indeed proposes double aromatic character to the former. In the case of C4N systems, both the NICS values are higher for C12 than for C8 and a steady decrease with further increase in size. Although this may indicate increasing stabilization of larger rings, the size dependency of NICS has to be accounted to get the true effect. Hence, we compare the NICS(0) of C8 (44.21) with that of similar sized ring system cyclooctatetraene NICS(0) (30.1 (ref. 54)) and proposes that the former has significant double antiaromatic character.
Study of intermolecular interactions: formation of dimers
The nature of intermolecular interactions of the C4N+2 and C4N molecules are compared by studying their dimers. Fig. 5 displays the optimized geometries of the dimers of Cn containing 6 to 28 carbon atoms. In C4N+2 dimers, the monomers show a shifted-parallel stacked orientation whereas in C4N dimers, the monomers are seen in a perfect stacking arrangement. The C4N dimer structure can be described as a sandwich type configuration wherein the shorter CC bonds orient on top of the longer CC bonds. The centre-to-centre distances and the nearest C⋯C distances in the dimers are also given in Fig. 5. These distances are almost constant in C4N+2 dimers. The centre-to-centre distances show a slight decrease of 0.57 Å with increase in ring size from C8 (2.77 Å) to C28 (3.34 Å) in the case of C4N dimers. The nearest C⋯C distances also possess similar values in them. The QTAIM plots of dimers of C4N+2 and C4N molecules are given in Fig. 6 and it clearly shows that in C4N dimers, the monomers are connected together by a larger number of C⋯C interactions compared to C4N+2 dimers. The interactions shown by dotted lines in Fig. 5 represent bond paths and are characterized by a bond critical point at the midpoint region. Except for C8, the number of C⋯C interactions in a C4N dimer is equal to the number of carbon atoms in a monomer.
 |
| | Fig. 5 Optimized geometry of (a) C4N+2 dimers and (b) C4N dimers. The centre-to-centre distances and the nearest C⋯C distances in the dimers are given in Å. | |
 |
| | Fig. 6 QTAIM plots of (a) C4N+2 dimers and (b) C4N dimers. Dotted lines indicate bond paths for the C⋯C interactions. | |
The MESP map of Cn dimers given in Fig. 7 can explain the difference in intermolecular bonding behaviour in C4N and C4N+2 rings. As shown previously, the monomers of C4N rings are clearly partitioned into electron rich and electron deficient regions. When these molecules form dimers, the electron rich regions on one monomer faces the electron deficient regions of the other to obtain maximum complimentary electrostatic interactions, which leads to large number of intermolecular C⋯C interactions. In C4N+2 rings, partitioning of the monomers into electron rich and electron deficient regions is not clearly demarcated due to double aromaticity and leads to fewer number of C⋯C interactions, lower interaction energy and lower tendency towards dimer formation compared to C4N systems.
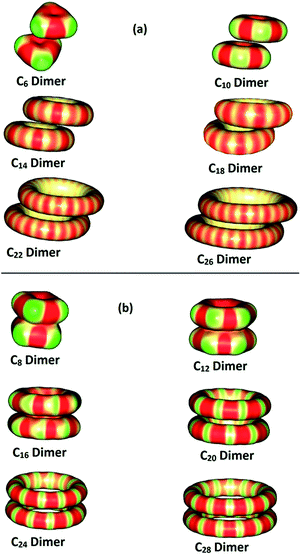 |
| | Fig. 7 MESP mapped on to 0.01 au isosurface of dimers of (a) C4N+2 and (b) C4N. Colour coding from blue to red indicates MESP values in the range −0.03 to 0.05 au. | |
The interaction energies (Eint) and average electron density at intermolecular bond critical points (ρave) of each of the Cn dimers are given in Table 3. In C4N+2 dimers, the magnitude of Eint shows a steady increase from C6 (−2.23 kcal mol−1) to C26 (−11.77 kcal mol−1). The ρave values are almost similar in all the C4N+2 dimers (a small variation between 0.0049 and 0.0057 au) indicating similar strength of intermolecular C⋯C interactions in all of them. On the other hand, in C4N dimers, the largest magnitude for Eint is shown by dimers of the smallest rings, viz. C8 (−23.31 kcal mol−1) and C12 (−21.80 kcal mol−1). The magnitude of Eint decreases up to C20 and then shows a steady increase for C24 and C28. This trend of Eint values is the result of a balance between the number of C⋯C interactions and the strength of individual C⋯C interactions. The ρave values show that C8 (ρave = 0.0179 au) and C12 (ρave = 0.0113 au) dimers possess the strongest C⋯C interactions as they show the highest amount of charge separation in terms of MESP features. The value of ρave gradually decreases from 0.0179 au in C8 to 0.0056 au in C28 as the ring size increases. The ρave value of C24, 0.0058 au is close to that of C28 and suggests that with further increase in ring size, the ρave value may not undergo substantial changes. Since the number of C⋯C interactions is equal to 4N in C4N dimers, an increase in the ring size beyond C28 is bound to increase the total interaction energy. The presence of a large number of intermolecular C⋯C interactions in C4N molecules also supports our previous studies35,36 that a clear demarcation of electron rich and electron deficient regions in molecules can result in intermolecular complimentary electrostatic interactions between even chemically similar atoms.
Table 3 Interaction energy (Eint) and average of electron density (ρave) at intermolecular BCPs corresponding to C⋯C interactions of C4N+2 and C4N dimers
| C4N+2 |
Eint (kcal mol−1) |
ρave (au) |
C4N |
Eint (kcal mol−1) |
ρave (au) |
| C6 |
−2.23 |
0.0053 |
C8 |
−23.31 |
0.0179 |
| C10 |
−5.20 |
0.0049 |
C12 |
−21.80 |
0.0113 |
| C14 |
−6.92 |
0.0057 |
C16 |
−15.83 |
0.0092 |
| C18 |
−8.32 |
0.0055 |
C20 |
−15.39 |
0.0061 |
| C22 |
−9.98 |
0.0049 |
C24 |
−16.88 |
0.0058 |
| C26 |
−11.77 |
0.0049 |
C28 |
−18.27 |
0.0056 |
Though the presence of a BCP may not always indicate a bonding situation,59,60 analysis of molecular orbitals (MOs) suggests strong orbital overlap between the two monomers in C4N dimers. Occupied MOs corresponding to the C⋯C interactions in C16 are given in Fig. 8 as a typical example for the C4N systems. On the other hand, C4N+2 systems show very few such MOs corresponding to C⋯C interactions. For e.g., C18 shows only one such MO (ESI†).
 |
| | Fig. 8 Occupied MOs corresponding to the inter-molecular C⋯C interactions in C16 plotted at 0.02 au isosurface. | |
Band gap of Cn systems
The HOMO–LUMO gap of Cn systems are given in Fig. 9. This gap in C4N+2 shows a steady decrease with increase in ring size suggesting increased degree of delocalization in the larger systems, except in the case of C6. In C6, the three inner lying carbons mutually interact at the CC distance 1.809 Å. These additional bonding interactions changes the nature of the HOMO and LUMO states compared to the other systems. The HOMO–LUMO gap value decreases from 4.31 eV in C10 to 1.44 eV in C30. Though small, a decrease in band gap is observed in C4N rings, on going from C8 (1.36 eV) to C32 (0.84 eV).
 |
| | Fig. 9 Band gap of C4N+2 and C4N molecules. | |
Homodesmotic reactions leading to polyynes
The stability of the cyclic Cn molecules has been assessed on the basis of energy of hypothetical homodesmotic reactions. In these reactions, a Cn ring reacts with acetylene to form a polyyne (Fig. 10). The hybridization state of all the atoms, number of CC bonds and number of CH bonds are conserved in the reaction. However, the aromatic/antiaromatic character of the ring and the associated strain effects are not conserved in the product side of the reaction. Therefore the energy of the reaction gives a direct estimate of the total stabilizing/destabilizing effect of the molecule from aromatic/antiaromatic character and strain in the ring structures.
 |
| | Fig. 10 Hypothetical homodesmotic reaction between C18 and acetylene to form linear C20H2 (10yne). | |
Table 4 gives the energy of the homodesmotic reactions (Ehdr) for C4N+2 and C4N rings. All the reactions are exothermic suggesting that the ring systems are less stable than the linear configurations. For smaller rings, the reaction is expected to be more exothermic due to larger strain effects. The energy released in the reaction (Ehdr) could be used as a measure of the stability of the ring structure with respect to the stability of the linear structure. If ring strain is the only effect contributing to the relative stability of these cyclic molecules, one would expect the highest exothermicity in the smallest ring, C6. However, from the Table 4, it is clear that the reaction of C8 is more exothermic by 16.5 kcal mol−1 compared to that of C6. Similarly, the reactions of the C4N systems, viz. C12, C16 and C20 with acetylene are more than 20 kcal mol−1 exothermic compared to those of the smaller sized C4N+2 systems, viz. C10, C14 and C18 respectively. In fact, Ehdr of C10 is comparable to that of C16. The C4N appears to be far more unstable than C4N+2 as the former is doubly antiaromatic while the latter is doubly aromatic. For both the sets, the magnitude of Ehdr decreases as the ring size increases, which can be attributed to decrease in ring strain with increase in ring size. Except in the case of the smallest rings (i.e., C6 and C8), the difference in Ehdr between each set of C4N+2 and the next higher C4N are lowered compared to the preceding set. For instance, the difference in Ehdr between C10 and C12 is 28.39 kcal mole−1, between C14 and C16 is 25.99 kcal mol−1 and between C30 and C32 is 9.88 kcal mol−1. This observation supports the assumption that at sufficiently large size of the ring, both kinds of molecules may show non aromatic character. The difference in the value of Ehdr between C6 and C8 is very small compared to that between C10 and C12, which can be attributed to the instability caused by high ring strain in C6. The results show that these ring systems are thermodynamically less stable compared to polyyne molecules.
Table 4 Energy of formation of polyynes from Cn rings and acetylene (Ehdr, in kcal mol−1)
| Reactants |
Product |
Ehdr |
Reactants |
Product |
Ehdr |
| C6 + C2H2 |
4yne |
−130.01 |
C8 + C2H2 |
5yne |
−146.54 |
| C10 + C2H2 |
6yne |
−77.52 |
C12 + C2H2 |
7yne |
−105.91 |
| C14 + C2H2 |
8yne |
−55.35 |
C16 + C2H2 |
9yne |
−81.34 |
| C18 + C2H2 |
10yne |
−41.74 |
C20 + C2H2 |
11yne |
−62.33 |
| C22 + C2H2 |
12yne |
−33.27 |
C24 + C2H2 |
13yne |
−49.35 |
| C26 + C2H2 |
14yne |
−27.55 |
C28 + C2H2 |
15yne |
−40.14 |
| C30 + C2H2 |
16yne |
−23.53 |
C32 + C2H2 |
17yne |
−33.41 |
Opening up of carbon rings to linear carbon chains
A Cn carbon ring is isomeric to a linear Cn carbon chain. The former with n C–C bonds is expected to be more stable compared to the latter with n − 1 C–C bonds. The ring structure experiences destabilizing ring strain effect. Aromaticity is another factor that controls the stability. Therefore, the energy of a reaction (Eopening) that addresses the opening up of the ring structure of a C4N or C4N+2 system to the corresponding chain structure (Fig. 11) is very useful to compare their stability.
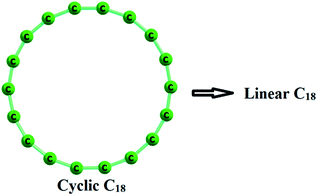 |
| | Fig. 11 Opening of cyclic C18 to linear C18. | |
In the case of C4N+2 rings, the extra stabilization due to aromaticity will be lost while in C4N, the destabilizing antiaromaticity will disappear due to ring opening. Therefore, opening up of a C4N+2 ring will be thermodynamically more difficult compared to that of a C4N ring. In both the cases, the ring strain effect will work in favour of the forward direction of the reaction. Eopening of both C4N and C4N+2 rings are given in Table 5. This data suggests that as the ring size increases, the stability of the ring increases which is graphically illustrated in Fig. 12. In the case of C6, the ring opening is endothermic by 22.8 kcal mol−1 while the value of Eopening of C8 is only 0.8 kcal mol−1. Similarly all the smaller rings, viz. C10, C14, C18, C22, C26 and C30 with C4N+2 character give higher endothermic reactions than the larger rings with C4N configuration, viz. C12, C16, C20, C24, C28 and C32, respectively. In fact, if C4N behave similar to C4N+2, the expected Eopening would be significantly higher (the data labelled using white squares in Fig. 12) than the actual values. Similarly, if C4N+2 behave like C4N, the expected Eopening would be much lower (the data labelled using the white circles). The ‘aromatic’ and ‘antiaromatic’ curves plotted in Fig. 12 are useful to derive a ‘non aromatic’ curve which is assumed to pass through the mid region of these two curves. This assumption is helpful to make a rough estimate about aromatic stabilization of a C4N+2 ring or the antiaromatic destabilization of C4N ring. The data in Fig. 12 suggest that C6, C10, C14, C18, C22, C26, and C30 are stabilized by 21.9, 23.3, 18.1, 14.5, 11.0, 8.1 and 6.7 kcal mol−1 due to aromaticity while C8, C12, C16, C20, C24, C28 and C32 are destabilized by 22.7, 21.1, 17.0, 12.7, 9.7, 7.3 and 5.6 kcal mol−1 due to antiaromaticity. This data further indicate that as the ring size increases (one exception is C10), stabilization/destabilization due to aromaticity/antiaromaticity decreases and the energetics of the system may favour a non aromatic state at sufficiently large size. However, the NICS values support this conclusion only for C4N systems. For C4N+2 systems, these values indicate highly aromatic character even at larger sizes.
Table 5 The energy required for ring opening (Eopening) in kcal mol−1 of C4N and C4N+2 rings
| C4N+2 |
Eopening |
C4N |
Eopening |
| C6 |
22.84 |
C8 |
00.79 |
| C10 |
66.38 |
C12 |
35.56 |
| C14 |
84.34 |
C16 |
57.08 |
| C18 |
95.66 |
C20 |
74.27 |
| C22 |
102.72 |
C24 |
86.17 |
| C26 |
107.62 |
C28 |
94.75 |
| C30 |
111.09 |
C32 |
101.05 |
 |
| | Fig. 12 The variation in Eopening with number of carbon atoms in aromatic and antiaromatic Cn rings. | |
Conclusions
Cn rings (from n = 6–32) with even number of carbon atoms have been studied for their aromatic and dimer formation features using the DFT method M06L. The geometric, electrostatic, magnetic and energetic features indicated strong aromatic stabilization in C4N+2 rings and strong antiaromatic character in C4N rings. The C4N+2 systems showed cumulenic structures with all the bond lengths being equal while the C4N rings possessed acetylenic structures with clear bond length alternation. The MESP features clearly indicated delocalization of π-electrons and aromaticity in C4N+2 rings while electron localization around the shorter CC bond (formal triple bond) in C4N rings as visualized from MESP proposed their antiaromaticity. The NICS analysis showed high negative values in C4N+2 and high positive values in C4N. The magnitude of these values was much higher than similar sized aromatic hydrocarbons and indicated double aromatic/antiaromatic features of the corresponding Cn rings. With increase in the ring size, the magnitude of NICS in a C4N+2 ring increased while those of C4N ring decreased, both indicating stabilization, which is attributed mainly to the decrease in ring strain effects. Hypothetical homodesmotic reactions and ring opening reactions have been used to quantify the higher stability of C4N+2 rings over C4N rings. The Cn rings are thermodynamically less stable compared to the corresponding polyyne systems which is accounted by the ring strain effect in the former. As the ring size increases, the destabilizing strain effect decreases and the stability of a Cn ring approaches close to that of a polyyne. In fact, the aromatic C4N+2 systems such as C14, C18, C22, C26 and C30 possessing a smooth MESP distribution should be targeted for synthesis.
The difference in geometric features has a clear effect in the dimer formation behaviour of C4N+2 and C4N rings. The dimer of C4N+2 always showed significantly lower magnitude of Eint compared to the dimer of a C4N ring. C4N+2 with equal bond lengths and a well delocalized system of π electrons have very low tendency to form dimers compared to C4N. The number and strength of intermolecular C⋯C interactions are also very less in C4N+2 dimers due to the lack of intermolecular complimentary electrostatic interactions. On the other hand, the very high magnitude of Eint observed for C4N could be correlated to the large number (equal to the number of carbon atoms in the monomer, except for C8) of inter-molecular C⋯C interactions as seen in the QTAIM analysis. These interactions, resulting from complimentary electrostatic interactions between electron rich formal triple bond regions of one monomer with the relatively electron deficient region of the second give rise to perfect stacking sandwich type arrangement of the monomers in a C4N dimer. The fact that the C4N dimers possess large number of C⋯C interactions supports our previous studies35,36 that separation of electron rich and electron deficient regions in a molecule can result in the formation of intermolecular C⋯C bonding interaction between atoms in similar chemical environments.
Acknowledgements
This research is supported by the Council of Scientific and Industrial Research (CSIR), Govt. of India, through a project CSC0129. K. R. is thankful to CSIR, India, for providing a senior research fellowship.
References
- J. Hunter, J. Fye and M. F. Jarrold, Science, 1993, 260, 784–786 CAS.
- R. O. Jones, J. Chem. Phys., 1999, 110, 5189–5200 CrossRef CAS.
- S. Tongay, S. Dag, E. Durgun, R. T. Senger and S. Ciraci, J. Phys.: Condens. Matter, 2005, 17, 3823–3836 CrossRef CAS PubMed.
- A. Van Orden and R. J. Saykally, Chem. Rev., 1998, 98, 2313–2358 CrossRef CAS PubMed.
- J. Hutter, H. P. Luethi and F. Diederich, J. Am. Chem. Soc., 1994, 116, 750–756 CrossRef CAS.
- G. von Helden, M. T. Hsu, N. Gotts and M. T. Bowers, J. Phys. Chem., 1993, 97, 8182–8192 CrossRef CAS.
- S. Irle, G. Zheng, Z. Wang and K. Morokuma, J. Phys. Chem. B, 2006, 110, 14531–14545 CrossRef CAS PubMed.
- W.-H. Lin, C.-C. Tu and S.-L. Lee, Int. J. Quantum Chem., 2005, 103, 355–368 CrossRef CAS.
- K. M. Rama, Y.-T. Lin and S.-L. Lee, Int. J. Quantum Chem., 2001, 84, 642–648 CrossRef.
- J. M. Hunter, J. L. Fye, E. J. Roskamp and M. F. Jarrold, J. Phys. Chem., 1994, 98, 1810–1818 CrossRef CAS.
- H. Schwarz, Angew. Chem., Int. Ed., 1993, 32, 1412–1415 CrossRef.
- N. S. Goroff, Acc. Chem. Res., 1996, 29, 77–83 CrossRef CAS.
- G. von Helden, M. T. Hsu, P. R. Kemper and M. T. Bowers, J. Chem. Phys., 1991, 95, 3835–3837 CrossRef CAS.
- R. O. Jones and G. Seifert, Phys. Rev. Lett., 1997, 79, 443–446 CrossRef CAS.
- J. M. Hunter, J. L. Fye and M. F. Jarrold, J. Chem. Phys., 1993, 99, 1785–1795 CrossRef CAS.
- T. Giesen, A. Van Orden, H. Hwang, R. Fellers, R. Provencal and R. Saykally, Science, 1994, 265, 756–759 CAS.
- S. Tongay, R. T. Senger, S. Dag and S. Ciraci, Phys. Rev. Lett., 2004, 93, 136404 CrossRef CAS PubMed.
- F. A. Fernandéz-Lima, C. R. Ponciano, E. F. da Silveira and M. A. C. Nascimento, Chem. Phys. Lett., 2007, 445, 147–151 CrossRef.
- K. Raghavachari and J. S. Binkley, J. Chem. Phys., 1987, 87, 2191–2197 CrossRef CAS.
- K. B. Shelimov, J. M. Hunter and M. F. Jarrold, Int. J. Mass Spectrom. Ion Processes, 1994, 138, 17–31 CrossRef CAS.
- G. von Helden, N. G. Gotts, P. Maitre and M. T. Bowers, Chem. Phys. Lett., 1994, 227, 601–608 CrossRef CAS.
- N. G. Gotts, G. von Helden and M. T. Bowers, Int. J. Mass Spectrom. Ion Processes, 1995, 149–150, 217–229 CrossRef.
- V. A. Schweigert, A. L. Alexandrov, Y. N. Morokov and V. M. Bedanov, Chem. Phys. Lett., 1995, 238, 110–115 CrossRef CAS.
- T. Wakabayashi, M. Kohno, Y. Achiba, H. Shiromaru, T. Momose, T. Shida, K. Naemura and Y. Tobe, J. Chem. Phys., 1997, 107, 4783–4787 CrossRef CAS.
- E. J. Bylaska, J. H. Weare and R. Kawai, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, R7488–R7491 CrossRef CAS.
- M. Saito and Y. Okamoto, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 8939–8942 CrossRef CAS.
- T. Torelli and L. Mitas, Phys. Rev. Lett., 2000, 85, 1702–1705 CrossRef CAS PubMed.
- V. I. Minkin, M. N. Glukhovtsev and B. I. A. Simkin, Aromaticity and antiaromaticity: electronic and structural aspects, J. Wiley & Sons, 1994 Search PubMed.
- J. Chandrasekhar, E. D. Jemmis and P. von Ragué Schleyer, Tetrahedron Lett., 1979, 20, 3707–3710 CrossRef.
- E. J. Bylaska, R. Kawai and J. H. Weare, J. Chem. Phys., 2000, 113, 6096–6106 CrossRef CAS.
- S.-h. Xu, M.-y. Zhang, Y.-y. Zhao, B.-g. Chen, J. Zhang and C.-C. Sun, Chem. Phys. Lett., 2006, 421, 444–447 CrossRef CAS.
- M. D. Wodrich, C. Corminboeuf, S. S. Park and P. v. R. Schleyer, Chem.–Eur. J., 2007, 13, 4582–4593 CrossRef CAS PubMed.
- P. W. Fowler, N. Mizoguchi, D. E. Bean and R. W. A. Havenith, Chem.–Eur. J., 2009, 15, 6964–6972 CrossRef CAS PubMed.
- S. Sen, P. Seal and S. Chakrabarti, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 245401 CrossRef.
- K. Remya and C. H. Suresh, Phys. Chem. Chem. Phys., 2015, 17, 18380–18392 RSC.
- K. Remya and C. H. Suresh, Phys. Chem. Chem. Phys., 2015, 17, 27035–27044 RSC.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- K. Remya and C. H. Suresh, J. Comput. Chem., 2013, 34, 1341–1353 CrossRef CAS PubMed.
- N. Mohan and C. H. Suresh, Int. J. Quantum Chem., 2014, 114, 885–894 CrossRef CAS.
- K. Remya and C. H. Suresh, J. Comput. Chem., 2014, 35, 910–922 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. FoxGaussian 09, Revision C.01, Gaussian, Inc.: Wallingford CT, 2010 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. R. Gadre and R. N. Shirsat, Electrostatics of Atoms and Molecules, Universities Press, Hyderabad, India, 2000 Search PubMed.
- J. S. Murray, K. Paulsen and P. Politzer, Proc.–Indian Acad. Sci., Chem. Sci., 1994, 106, 267–275 CAS.
- P. Sjoberg and P. Politzer, J. Phys. Chem., 1990, 94, 3959–3961 CrossRef CAS.
- T. Clark, M. Hennemann, J. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- J. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 723–729 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, A Tool for Interpretation and Prediction. From Molecular Structure to Solvation Effects, in Molecular Electrostatic Potentials: Concepts and Applications, Elsevier, Amsterdam, 1996 Search PubMed.
- S. R. Gadre, P. Bhadane, S. S. Pundlik and S. S. Pingale, Molecular Electrostatic Potentials: Concepts and Applications, in Molecular Electrostatic Potentials: Concepts and Applications, Elsevier, Amsterdam, 1996 Search PubMed.
- S. R. Gadre and S. S. Pundlik, J. Phys. Chem. B, 1997, 101, 3298–3303 CrossRef CAS.
- A. Kumar, S. R. Gadre, N. Mohan and C. H. Suresh, J. Phys. Chem. A, 2014, 118, 526–532 CrossRef CAS PubMed.
- T. A. KeithAIMAll (Version 14.04.17); TK Gristmill Software, Overland Park KS, USA, 2014 Search PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- J. Poater, M. Duran, M. Solà and B. Silvi, Chem. Rev., 2005, 105, 3911–3947 CrossRef CAS PubMed.
- F. Feixas, E. Matito, J. Poater and M. Sola, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- P. v. R. Schleyer, H. Jiao, N. J. R. v. E. Hommes, V. G. Malkin and O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- C. Foroutan-Nejad, S. Shahbazian and P. Rashidi-Ranjbar, Phys. Chem. Chem. Phys., 2010, 12, 12630–12637 RSC.
- J. Poater, M. Solà and F. M. Bickelhaupt, Chem.–Eur. J., 2006, 12, 2889–2895 CrossRef CAS PubMed.
- J. Poater, M. Solà and F. M. Bickelhaupt, Chem.–Eur. J., 2006, 12, 2902–2905 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06833b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.